





















 このページを印刷する
このページを印刷する|
魚崎 英毅 先生 自治医科大学 分子病態治療研究センター 再生医学研究部 |
記事ID : 43775
QuantSeq 3'mRNA-Seq Library Prep Kitを用いたトランスクリプトーム解析
ユーザーレポート
Product
メーカー:Lexogen GmbH メーカー略号:LEX
■ QuantSeq 3' mRNA-Seq Library Prep Kit (FWD)
イルミナ社機器に対応!
網羅的遺伝子発現解析用ライブラリ調製キット

- 網羅的な遺伝子発現解析にお勧め
- 4.5 時間で total RNA から 3' mRNA-Seq ライブラリを調製可能
- マイクロアレイ解析や従来の RNA シークエンス解析と比較し低コスト
- 無料のデータ解析パイプライン「Bluebee®」をご用意
- Globin blockやマルチプレックス解析用インデックス、増幅バイアス確認用インデックス(UMIs)と組み合わせてライブラリ調製可能
- Unique Dual Indices(UDIs)とのセット品もご用意
実験内容
かつてトランスクリプトーム解析といえばマイクロアレイであったが、超並列シーケンサー(Massive Parallel Sequencer, MPS)の発展により、すっかりその座はRNAシーケンス(RNA-seq)に取って代わられた。超並列シーケンサーでは、同時に数百万から数十億のシーケンス反応を並行して行い、長きに渡りシーケンサー界で不動の地位を築いていたサンガーシーケンサーに対し、次世代シーケンサー(Next Generation Sequencer, NGS)とも呼ばれる。RNA-seqでは、細胞や組織からRNAを抽出し、逆転写したものをMPSによりシーケンスを行うことで網羅的に配列情報を得る。得られたそれぞれの配列を由来となった種の参照ゲノム配列や遺伝子産物の配列と一致する場所を探す(マップする)ことで、どの遺伝子産物がどれくらい発現しているかをマップされた数(カウント)として得ることができる。広く使われているRNA-seqではリボゾームRNAの除去やオリゴdTビーズを用いたポリAが付加したmRNAを濃縮し、その後、ランダムプライマーを使って逆転写したcDNAを用いる。これにより、mRNA全長をマップし、mRNAの発現だけでなく、転写産物のスプライシングなどまで解析することができる。1サンプルを解析するために必要な配列(リード)数は1,000万から3,000万と言われている。最近のシーケンサーであれば、数十億から百億のオーダーまで読めるようになっており、多検体の解析にも対応できるが、そういった最新鋭の機器を保有している研究機関は多くない。
我々の研究室で、RNA-seqを立ち上げた検討した際、経時的トランスクリプトームや様々な薬剤処理・遺伝的な操作に対する細胞の応答を解析しようと考えていたため、同時に数十〜数百検体の解析が必要となる見込みであった。また、学内でNextSeq550(Illumina社)が自由に使える環境であった。NextSeq550では最大4億リードが得られる機器であるため、1サンプルあたり3,000万リードと概算した場合、同時に解析できるサンプル数は12程度と、必要なデータを得るためには相当数シーケンスを行う必要があると想定していた。そんな中コスモ・バイオのカタログで出会ったのが、Lexogen社のQuantSeq 3’ mRNA-Seq Library Prep Kit(以下QuantSeq)である。QuantSeqではオリゴdTプライマーを用いてtotal RNAから逆転写を行い、さらにランダムプライマーで2本鎖cDNAを作製する(図1)。つまり、mRNAの精製・濃縮ステップもなく、簡便な操作だけでmRNAの3’末端のみのcDNAライブラリができる。これによりmRNAの全長からcDNAを作製するのに対し、大幅なリード数削減が期待され、100万〜1,000万リードで十分である(図2)。キットには96種類のバーコード配列(最近ではデュアルインデックスを使い384まで対応)が同梱されており、我々の研究室では同時に48〜96サンプルからライブラリを作製し、NextSeqを使ってシーケンスを行っている[Chanthra, Sci Rep, 2020; Miyamoto, Cell Rep Methods, 2021]。
3’末端のみをシーケンスすることで転写産物のカウンティングの定量性も高く、遺伝子長で補正する必要もないため、解析がシンプルである。また、試薬・シーケンスコストが低く抑えられ、1サンプルあたり1万円以下(QuantSeqおよびIlluminaのシーケンスキットの価格をサンプル数96で割った場合。人件費やその他必要なチップなど消耗品代は別)でトランスクリプトームが得られるなど非常に魅力的である。一方、スプライシングなどの解析には向いておらず、目的に応じた使い分けが必要となる。
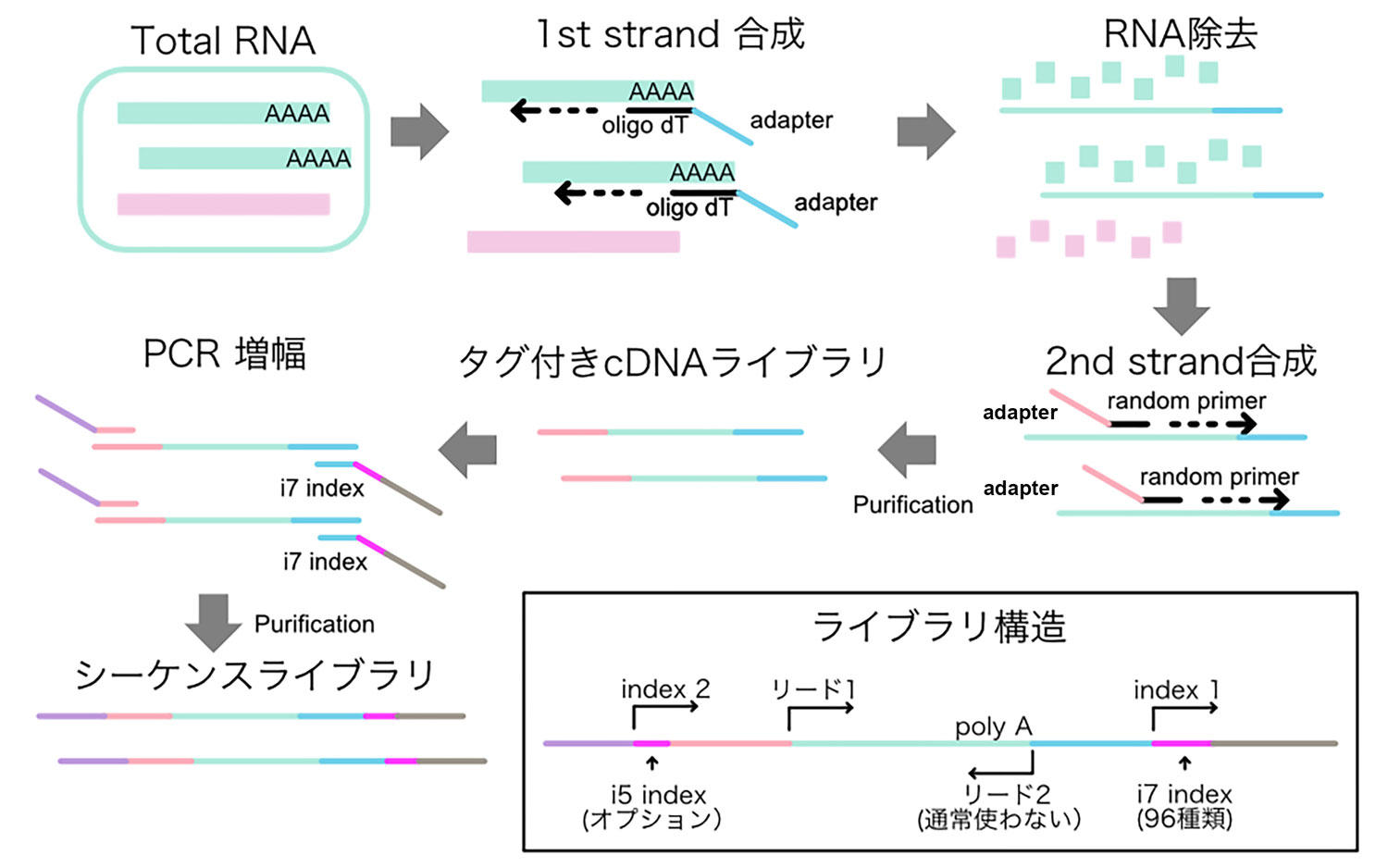
図1 ライブラリ作製の流れ
Total RNAのうち、ポリA配列を持つmRNAが選択的にoligo dTプライマーにより逆転写される。RNA除去後、ランダムプライマーにより2nd strandが合成される。以上の2種類のプライマーにはPCR増幅用の配列が付加されており、PCR増幅の際にi5/i7 indexが追加される。本キットにはi7 indexだけで96種類が添付されており、96種類のサンプルを同時にシーケンスすることができる。通常のシーケンスではリード1のみを用いるシングルエンドでシーケンスを行う。Lexogen社に問い合わせたところ、MiSeqでは50塩基読むSE50を、NextSeqでは75塩基読むSE75を推奨しており、ユニークマップ率を上げたければ、もう少し長く100-150塩基程度シーケンスすると良いとの回答を得ている。
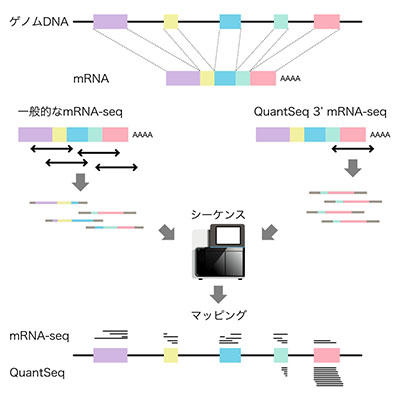
図2 一般的なmRNA-seqとQuantSeq 3’ mRNA-seqの比較
一般的なmRNA-seqの場合、mRNA全域をカバーするようなライブラリが作られる。そのため、検出されるエクソンからスプライシングバリアントのレベルで評価することができる。一方で、十分な定量性を得るにはシーケンスリード数が多く必要である。また、mRNA一分子あたりの塩基数は、mRNAの長さに依存するため、長いmRNAほど検出されやすくなる。そのため遺伝子発現解析を行う場合は、mRNAの長さによる補正を行う必要がある。QuantSeq 3’ mRNA-seqの場合、ライブラリはmRNAの3’末端に偏るため、リード数が少なくとも定量性が保たれる。また、長さによる補正も必要ないといった特徴がある。一方、スプライシングが検出できないこと、ユニークマップ率が低くなりやすいといった特徴もある。