





















- サービス詳細:Genome-CRISP™ Custom CRISPR-Cas9 受託サービス
- 商品詳細:GeneCopoeia社 CRISPR-Cas9 ノックアウトシステム
- 商品詳細:GeneCopoeia社 CRISPR-Cas9 HR(HDR)用ドナーベクター
- 商品詳細:GeneCopoeia社 CRISPR-Cas9 ノックインキット & HDR ノックインクローン
- 商品詳細:GeneCopoeia社 Genome-CRISP™ CRISPR-Cas9 stable cell line
- 商品詳細:GeneCopoeia社 Genome-CRISP™ human single guide RNA(sgRNA)ライブラリー
- 商品詳細:GeneCopoeia社 TALEN / CRISPR-Cas9 挿入欠損検出システム
■ ゲノム編集全般に関する質問
- 【01】 ゲノム編集技術を用いて点変異を導入できますか。
- 【02】 なぜドナー(HR, HDR)をゲノム編集に利用する必要があるのでしょうか。
- 【03】 GCP 社の商品やサービスはどのような生物種に適用可能ですか?
- 【04】 gRNAおよびCas9とドナー(HR, HDR)を同時にトランスフェクションする必要がありますか。
- 【05】 プラスミドは染色体に組み込まれますか?
- 【06】 CRISPR-Cas9システムのプラスミド(gRNA, Cas9)はどのような方法で細胞に送達すればよいですか。
- 【07】 選択マーカーがない場合の変異検出はどのように行えばよいのでしょうか?
- 【08】 セーフハーバーへの組換えや相同組換え(HR、HDR)を用いたゲノム編集が成功したかどうかをどのように検証すれば良いのでしょうか。
- 【09】 CRISPR-Cas9 システムは、希望のゲノム編集のあとも継続して染色体を切断し続けるのですか。
- 【10】 二対立遺伝子修正も可能でしょうか。
- 【11】 ゲノム編集において、TALENとCRISPR-Cas9システムの違いは何ですか。
- 【12】 TALENとCRISPR-Cas9のどちらを利用するべきでしょうか。
■ CRISPR-Cas9に関する質問
- 【13】 野生型Cas9 の代わりにニッカーゼ改変型Cas9を使用できますか?
- 【14】 ダブルニッキング法でドナー(HR、HDR)を利用できますか。
- 【15】 GCP社のCRISPR-Cas9 ノックアウトシステムとして提供されている各ベクターを、空ベクターの状態で購入することはできますか?
- 【16】 自分でドナー(HR、HDR)を構築するため、空のドナープラスミドを購入できますか。
- 【17】 CRISPR-Cas9技術を遺伝子活性化や抑制に利用できますか。
- 【18】 レンチウイルスベクターを利用したCRISPR-Cas9システムを提供していますか。
- 【19】 相同組換え(HR、HDR)にレンチウイルスを利用した送達法を使用できますか。
■ セーフ・ハーバーシステム
■ gRNA(sgRNA、短鎖RNA)ライブラリー
ゲノム編集全般に関する質問
可能です。変異を導入したいいずれか側の座位に相同するドナーが必要です。プラスミド、または一本鎖オリゴヌクレオチドのいずれもドナーとして利用できます。ドナーをゲノム編集ツール(CRISPR)と同時にトランスフェクションすると、二本鎖切断(DSB)が形成されることでドナーを利用した相同性組換え(HR、HDR)による切断部位修復が誘導されます。GCP社では、お客様があらかじめ決定した修正を含み、かつ、ピューロマイシン耐性やcopGFPなどの選択マーカーが隣接、そして標的領域に相同な側腕(ホモロジーアーム)をもつドナープラスミドのカスタム構築サービスを提供しています。マーカーは loxP 部位に隣接しているため Cre により除去することができます。ドナープラスミドを利用することで編集済みクローンを選択することができ、スクリーニングするクローンの数を低減できます。
ゲノム編集にドナー(HR、HDR)を利用する理由として主に3つの重要な点があげられます。
- 正確性が格段に向上し、かつ制御可能であること。
- どのような「期待する変更」をも実行可能であること。
- ノックインしたマーカー(薬剤耐性や蛍光)選択を利用できること。これにより期待する修正が生じた候補クローンの同定作業をスピードアップすることができる。
より詳細な情報は、ドナーを利用した効果的なゲノム編集アプリケーションをご参照ください。
全ての生物種に適用可能です。CRISPR-Cas9ノックアウトプラスミドは哺乳類細胞で機能するようデザインしています。さらに、T7 プロモーター駆動型in vitro転写法の鋳型として利用できます。CRISPR RNA を線虫やショウジョウバエ、ゼブラフィッシュ等にインジェクションすることが可能です。
はい。ドナー(HR、HDR)が修復鋳型として利用されるためには、DNA二本鎖切断(DSB)が形成した時点でドナー(HR、HDR)が存在している必要があります。未修復のDNA二本鎖切断(DSB)は細胞にとって致死的であるため、ドナーが存在しない場合には、非相同性末端結合(NHEJ)を利用してDNA二本鎖切断(DSB)を修復します。
CRISPR-Cas9 プラスミドをトランスフェクションする実験系では、通常、宿主ゲノムに組込まれません。ただし、ゲノム編集した細胞クローンの選択後、ヌクレアーゼプラスミドを欠損したクローンをスクリーニングすることをおすすめします。プラスミドにコードされていた耐性遺伝子の抗生物質に感受性であるか、または、プラスミドにコードされていた蛍光マーカーを発現しないかなどを確認する方法が利用できます。レンチウイルスクローンは染色体上にランダムに組込まれます。
より詳細な情報は、CRISPR-Cas9 - 実験計画を立てる!をご参照ください。
CMVやEF1aプロモーターが存在する場合、プラスミドDNAからの発現が生じます。対象とする細胞種において最も効果の高い手法を用いることをおすすめします。GCP社のゲノム編集ツールを細胞に送達する際に利用できる手法は以下です。
- 標準的なトランスフェクション法
- エレクトロポレーション
- レンチウイルスのトランスダクション
- DNAまたはmRNAの微量注入法
in vitro転写法によりmRNAを産生可能なT7プロモーターを含むプラスミド
より詳細な情報は、CRISPR-Cas9 - 実験計画を立てる!をご参照ください。
相同性ドナー(HR、HDR)を使用せず、を用いずに CRISPR による標的遺伝子の変異を行った場合、編集されたクローンを同定するために多くの作業が必要になります。トランスフェクション後、単一細胞より生長した数多くのクローンを単離します。以下に示すように、スクリーニング工程はどのようなゲノム編集を実施したかによって異なります。
- 挿入や欠損を導入する場合、最も簡便なスクリーニング方法は修正部位を挟んだ位置に設計したプライマーを用いてPCR増幅することです。ただし、挿入または欠損が一般的なアガロースゲル電気泳動法で検出可能な大きさである必要があります。
- 挿入や欠損が非常に小さい場合、クローンのスクリーニングにはミスマッチを含む二本鎖DNAを切断することで変異を検出するT7エンドヌクレアーゼI(Surveyor)アッセイが利用できます。リアルタイムPCRを利用することもできます。 GeneCopoeiaでは、挿入欠損検出用のT7エンドヌクレアーゼI切断アッセイキットをご提供しています。
- 点変異を導入する場合、修正検出にはSurveyorアッセイまたはリアルタイムPCRが利用できます。
- ゲノム編集を実施したことによって、制限酵素サイトが産生または破壊される場合、PCR 産物の酵素切断により修正済みまたは未修正の対立遺伝子を区別することができます。
- PCR産物のサンガーシークエンシングや全ゲノムの次世代型シークエンシングを利用して修正を確認する方法もあります。
いずれのスクリーニング手法を選択しても、一部分のみが修正したのか、全対立遺伝子が修正したのかを同定できることが重要です。 編集されたクローンを同定するために要する時間と労力を削減するために、GCP 社のドナープラスミドカスタム構築サービスをおすすめ致します。
本サービスでは、標的配列に相同なアームとピューロマイシン耐性等の選択マーカー、copGFP を含むドナープラスミドをデザイン・構築します。マーカーは loxP に隣接しているため Cre により除去することができます。
セーフ・ハーバーへの組込み検証には、ランダムな組込みを除外できるよう、組換え座位を増幅するようデザインしたプライマーを使用してPCRを行うか、サザンブロッティングにより検証できます。全てのGCP 社の Genome TALER Human AAVS1 Safe Harbor Gene knock-in Kitには検証のための PCR プライマーペアが含まれています。相同組換え(HR、HDR)を用いた他のゲノム編集実験にも編集部位同定にPCRが利用できます。PCR法では、野生型と修正座位におけるPCR産物サイズの変化が確認できます。修正座位において期待するサイズの単一バンドが得られた場合、全コピーにおいて修正されたことが示唆されます。もし2本のバンドが得られた場合、少なくとも1つの対立遺伝子は未修正であることが示唆されます。サザンブロッティングもランダム組込みを検出するために有効です。
GCP社のCRISPR-Cas9ベクター骨格は、宿主ゲノムに組込まれたり、複製されないようにデザインされています。一過性に導入されたプラスミドは、通常、数回の細胞分裂後に消失し、宿主細胞にそれ以上影響を及ぼしません。トランスフェクション後、細胞を低密度で播種して単一コロニー形成させ、これらのコロニーをスクリーニングしてプラスミドが消失したことを確認します。プラスミドにコードされていた耐性遺伝子の抗生物質に感受性であるか、あるいは、プラスミドにコードされていた蛍光マーカーを発現しないかなどを確認する方法が利用できます。確実な一過的トランスフェクションにはプラスミド DNA の代わりに mRNA のご使用を推奨致します。
はい。CRISPR-Cas9では、複数コピーを同時に破壊できることが示されています。ただし、細胞種、トランスフェクション効率、CRISPR活性など、様々な要因によりその効率は異なります。
TALENとCRISPR-Cas9のいずれのシステムでも、標的部位特異的にデザインしたゲノム編集ツールが標的配列を認識し、ゲノム編集が惹起されます。最も大きな違いは、それがどのように生ずるかです。CRISPR-Cas9では、標的配列に相補性をもつ20塩基の単鎖ガイドRNA(gRNA, sgRNA)を利用します。この標的配列には直後にプロトスペーサー隣接モチーフ(PAM)とよばれる3塩基(N-G-G)配列が必要です。単鎖ガイドRNAがCas9ヌクレアーゼを希標的部位に誘導します。したがって、CRISPR-Cas9システムを使用する場合には、単鎖ガイドRNA配列をデザインする必要があります。
TALENは、34アミノ酸からなる反復配列の可変アミノ酸を利用して標的配列を認識するタンパク質です。この可変アミノ酸は反復可変二残基(Repeat Variable Diresidues:RVDs)として知られています。各反復配列には1つのRVDが含まれ、4種のRVDのそれぞれが特定の核酸塩基を認識します。
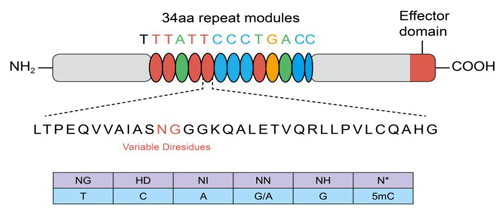
TALENは、ヌクレアーゼや転写修飾因子をはじめとするゲノム編集モチーフに融合します。TALEN技術を利用する場合、RDVsを標的遺伝子と一致するよう正しい順序に組み立てる必要があります。
より詳細な情報は、CRISPRとTALENの利点と欠点を解説をご参照ください。
非常に難しいご質問ですが、実験の最終目的により異なります。各システムには利点と欠点があります。CRISPR-Cas9は、TALENに比べて遺伝子編集効率が高い傾向にあります。また、CRISPR-Cas9はDNAメチレーションに感受性を示さずマルチプレックスをより行いやすいことからTALENよりも優位です。一方、CRISPR-Cas9は、ニッカーゼ改変型Cas9を含めオフターゲット部位を修正する傾向もあり、特定のアプリケーションによっては問題になることもあります。
より詳細な情報は、CRISPRとTALENの利点と欠点を解説をご参照ください。
CRISPR-Cas9に関する質問
はい。GCP社ではCas9 D10Aニッカーゼ変異体を含むプラスミドをご用意しており、GCP社のダブルニッカーゼデザインを用いた試験において、良好な結果を得ています。ただし、ダブルニッキング法によりDNA二本鎖切断(DSB)を行うためには、適切な間隔をもち、お互いが反対鎖に配置され、かつ切断場所を挟んだ2つのgRNA(sgRNA、ガイドRNA)が正確に標的する必要があります。さらに、適切なgRNA(sgRNA、ガイドRNA)標的には認識部位の直後に5'-NGG-3' "PAM"配列が存在する必要があり、どのような場合でもニッカーゼ改変型Cas9をDNA二本鎖切断に使用できるわけではありません。
より詳細な情報は、CRISPR-Cas オフターゲット作用をコントロールするをご参照ください。
利用できます。ダブルニッキング法によりDNA二本鎖切断(DSB)を行うためには、適切な間隔をもち、お互いが反対鎖に配置され、かつ切断場所を挟んだ2つのgRNA(sgRNA、ガイドRNA)が正確に標的する必要があります。この手法は非相同末端結合の発生率を減らすことによりオフターゲット切断のリスクを低下させることができます。ただし、適切なgRNA(sgRNA、ガイドRNA)標的には認識部位の直後に5'-NGG-3' "PAM"配列が存在する必要があり、どのような場合でもニッカーゼ改変型Cas9をDNA二本鎖切断に使用できるわけではありません。
より詳細な情報は、CRISPR-Cas オフターゲット作用をコントロールするをご参照ください。
デザインおよび構築済sgRNAコンストラクトのみを販売しており、空ベクターはご提供しておりません。ネガティブコントロールとして、弊社スクランブルsgRNAを組込んだCRISPRプラスミドをご提供しています。
商品の詳細情報は、CRISPR-Cas9 ノックアウトシステムをご参照ください。
はい、販売しています。
商品の詳細情報は、CRISPR-Cas9 HR(HDR)用ドナーベクターをご参照ください。
はい。Cas9ヌクレアーゼから、完全にヌクレアーゼ活性を消失させた二重変異体(dCas9)があります。この変異体をVP64などの転写修飾因子に融合させ、特定遺伝子標的に利用することができます。触媒作用を失活させたCas9と適切にデザインしたgRNA(sgRNA、短鎖RNA)を利用して遺伝子発現抑制や干渉を行うこともできます。
サンタクルズ社では、内在性の転写因子を強力に活性化するCRISPR-dCas Activation システムをご提供しています。
お問い合わせ先 : 創薬・受託サービス部
ご質問・ご不明の点は創薬・受託サービス部までお問い合せください。また、秘密保持契約のご希望につきましても、下記までご連絡をお願いいたします。
- TEL
- 03-5632-9615
- FAX
- 03-5632-9614
- jutaku_gr@cosmobio.co.jp
はい。非ウイルス系とレンチウイルス系の両方をご提供しています。レンチウイルス粒子作製サービスも承っており、Cas9とgRNA(sgRNA、短鎖RNA)の両方を発現するレンチウイルス粒子をお届け致します。
商品の詳細情報は、CRISPR-Cas9 ノックアウトシステムをご参照ください。レンチウイルス粒子作製サービスについては、以下にお問合せください。
残念ながらできません。レンチウイルスは細胞にRNAとして侵入しますが、HRドナーはCas9やgRNA(sgRNA、短鎖RNA)と同時にDNAとして細胞内に導入されなくてはなりません。
セーフ・ハーバーシステム
はい。GCP社では、ヒト(AAVS1)とマウス(Rosa26)を利用したCRISPR-Cas9セーフ・ハーバー組込みキットをご提供しています。これらのキットはGCP社のセーフ・ハーバーノックインORFクローンに対応しています。
商品の詳細情報は、CRISPR-Cas9 ノックインキット & HDR ノックインクローンをご参照ください。
gRNA(sgRNA、短鎖RNA)ライブラリー
レンチウイルス粒子、すぐにトランスフェクション可能なプラスミドDNA、およびバクテリアルストックの3種の形態をご用意しています。
商品の詳細情報は、Genome-CRISP™ human single guide RNA(sgRNA)ライブラリーをご参照ください。
はい。現在、GCP社のsgRNAライブラリはプール型式でご提供しています。もし、各クローンを個別に整列させたライブラリーをご希望の場合にもご対応させて頂けますので、以下にお問合せください。
お問い合わせ先 : 創薬・受託サービス部
ご質問・ご不明の点は創薬・受託サービス部までお問い合せください。また、秘密保持契約のご希望につきましても、下記までご連絡をお願いいたします。
- TEL
- 03-5632-9615
- FAX
- 03-5632-9614
- jutaku_gr@cosmobio.co.jp
はい。ただし、ライブラリー内の各gRNA(sgRNA、短鎖RNA)のレプリゼンテーションを一貫して維持するため、GCP社では、はじめにCas9安定発現細胞株を構築することを推奨しています。まず、Cas9レンチウイルスクローンを対象細胞にトランスダクションします。構築したCas9安定発現株に、gRNA(sgRNA、短鎖RNA)ライブラリーをトランスダクションします。
GCP社では、H1299、HEK293TおよびHeLa細胞のAAVS1位置にCas9を組み込んだCas9安定発現細胞株をご提供しています。
商品の詳細情報は、Genome-CRISP™ CRISPR-Cas9 stable cell lineをご参照ください。