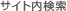






















CRISPR ‚â TALEN ‚ب‚اƒQƒmƒ€•زڈWƒcپ[ƒ‹‚ً—p‚¢‚½ژہŒ±‚إ‚حپA•W“I‚جچ×–E‚â“®•¨‚ھ“Kگط‚ة•زڈW‚³‚ê‚ؤ‚¢‚é‚©ٹm”F‚·‚邽‚ك‚جƒXƒNƒٹپ[ƒjƒ“ƒOژہŒ±‚âŒںڈطژہŒ±‚ھ•K—v‚ة‚ب‚邱‚ئ‚ھ‚µ‚خ‚µ‚خ‚ ‚è‚ـ‚·پBˆê”ت“I‚ة‚و‚ژg—p‚³‚ê‚ؤ‚¢‚éŒںڈطƒcپ[ƒ‹‚ئ‚µ‚ؤ “mismatch cleavage assay” ‚ھ‚ ‚è‚ـ‚·پBGeneCopoeiaژذ‚إ‚ح “mismatch cleavage assay” ‚ً—ک—p‚µ‚½ IndelCheck™ •دˆظŒںڈoƒLƒbƒg‚ً’ٌ‹ں‚µ‚ؤ‚¨‚èپAٹب’P‚ةƒQƒmƒ€•زڈW‚ة‚و‚é•دˆظ“±“ü‚جٹm”F‚ًچs‚ء‚ؤ‚¢‚½‚¾‚¯‚ـ‚·پB
‹Zڈpڈî•ٌ
‚ب‚؛ CRISPR ‚â TALEN ‚ً—p‚¢‚½ƒQƒmƒ€•زڈW‚ة‚ح‹@”\Œںڈط‚ھ•K—v‚ب‚ج‚©
‚ـ‚¸پACRISPR ‚â TALEN ‚ً—p‚¢‚ؤƒQƒmƒ€•زڈW‚ًچs‚¤چغ‚ة‚حپAژ–‘O‚ة sgRNA ‚جŒّ—¦‚ًŒںڈط‚·‚邱‚ئ‚ً‚¨ٹ©‚ك‚µ‚ـ‚·پBCRISPR ‚â TALEN ‚ة‚و‚éƒQƒmƒ€•زڈW‚حچ‚Œّ—¦‚ئچl‚¦‚ç‚ê‚ؤ‚¢‚ـ‚·‚ھپAƒ^پ[ƒQƒbƒg•”ˆت‚جگ«ژ؟‚ة‚و‚ء‚ؤ•زڈWŒّ—¦‚ح‘ه‚«‚•د‰»‚µ‚ـ‚·پB‚µ‚½‚ھ‚ء‚ؤپA•دˆظ“±“üŒمپA‰؛—¬‚جژہŒ±‚ةژg—p‚·‚é‘O‚ةپA‚ا‚جچ×–EƒNƒچپ[ƒ“‚ھچإ‚à“Kگط‚©‚ً•]‰؟‚·‚邱‚ئ‚ھڈd—v‚إ‚·پB
IndelCheck™ ƒVƒXƒeƒ€‚ح“ٌ–{چ½گط’f‚ً‰î‚µ‚½”ٌ‘ٹ“¯––’[Œ‹چ‡پiNHEJپj‚ة‚و‚èگ¶‚¶‚½ indelپi•دˆظ; ‘}“ü‚ـ‚½‚حŒ‡ژ¸پj‚ًŒںڈo‚·‚邽‚ك‚جƒZƒ‹ƒxپ[ƒXƒAƒbƒZƒCƒVƒXƒeƒ€‚إ‚·پBپiƒڈپ[ƒNƒtƒچپ[‚حگ}‚P‚ًژQڈئپj
گ}1پDIndelCheck™ ƒVƒXƒeƒ€‚جƒڈپ[ƒNƒtƒچپ[
CRISPR ‚ـ‚½‚ح TALEN ‚ة‚ؤƒQƒmƒ€•زڈW‚ًچs‚ء‚½چ×–E‚ة‚آ‚¢‚ؤپA•W“I•”ˆت‚ً‹²‚ٌ‚¾ˆت’u‚ةگفŒv‚µ‚½ƒvƒ‰ƒCƒ}پ[‚ً—p‚¢‚ؤPCR‚ًچs‚¤پBژں‚ةPCRژY•¨‚ً•دگ«ŒمپAچؤƒAƒjپ[ƒٹƒ“ƒO‚·‚邱‚ئ‚إپAƒ~ƒXƒ}ƒbƒ`‚ًٹـ‚ق“ٌ–{چ½ڈW’c‚ًŒ`گ¬‚³‚¹‚éپiƒzƒ‚“ٌ–{چ½پAƒwƒeƒچ“ٌ–{چ½پjپB‚±‚جƒ~ƒXƒ}ƒbƒ`‚ح T7 Endonuclease I ‚جٹîژ؟‚ئ‚ب‚èگط’f‚³‚ê‚éپBCRISPR ‚ـ‚½‚ح TALEN ‚ھچ×–E“à‚إ‹@”\‚µ‚½ڈêچ‡پAƒAƒKƒچپ[ƒXƒQƒ‹ڈم‚إگط’fژY•¨‚ًٹm”F‚·‚邱‚ئ‚ھ‚إ‚«‚éپB
CRISPR ‚ـ‚½‚ح TALEN ‚ة‚ؤƒQƒmƒ€•زڈW‚ًچs‚ء‚½چ×–E‚ة‚آ‚¢‚ؤپA•W“I•”ˆت‚ً‹²‚ٌ‚¾ˆت’u‚ةگفŒv‚µ‚½ƒvƒ‰ƒCƒ}پ[‚ً—p‚¢‚ؤPCR‚ًچs‚¤پBژں‚ةPCRژY•¨‚ً•دگ«ŒمپAچؤƒAƒjپ[ƒٹƒ“ƒO‚·‚邱‚ئ‚إپAƒ~ƒXƒ}ƒbƒ`‚ًٹـ‚ق“ٌ–{چ½ڈW’c‚ًŒ`گ¬‚³‚¹‚éپiƒzƒ‚“ٌ–{چ½پAƒwƒeƒچ“ٌ–{چ½پjپB‚±‚جƒ~ƒXƒ}ƒbƒ`‚ح T7 Endonuclease I ‚جٹîژ؟‚ئ‚ب‚èگط’f‚³‚ê‚éپBCRISPR ‚ـ‚½‚ح TALEN ‚ھچ×–E“à‚إ‹@”\‚µ‚½ڈêچ‡پAƒAƒKƒچپ[ƒXƒQƒ‹ڈم‚إگط’fژY•¨‚ًٹm”F‚·‚邱‚ئ‚ھ‚إ‚«‚éپB
IndelCheck™ ƒVƒXƒeƒ€‚جژg—p—ل
ڑM“û—قچ×–E‚إƒQƒmƒ€•زڈW‚ًچs‚¤ڈêچ‡پiin vitroپjپA“¯‚¶چ×–Eٹ”‚إŒںڈطژہŒ±‚ًچs‚¤‚±‚ئ‚ً‚¨ٹ©‚ك‚µ‚ـ‚·پB‚ـ‚½پAƒ}ƒEƒX‚⃉ƒbƒg‚ب‚اپAin vivo ‚إƒQƒmƒ€•زڈW‚ًچs‚¤ڈêچ‡‚حپAƒ}ƒEƒX‚إ‚ح Neuro2A چ×–E‚â NIH3T3 چ×–EپAƒ‰ƒbƒg‚إ‚ح PC-12 چ×–E‚ب‚ا‚جƒ‚ƒfƒ‹چ×–Eٹ”‚إŒںڈطژہŒ±‚ًچs‚¤‚±‚ئ‚ً‚¨ٹ©‚ك‚µ‚ـ‚·پBگ}‚P‚ةژ¦‚µ‚½‚و‚¤‚ةپAIndelCheck™ ƒVƒXƒeƒ€‚إŒںڈط‚ًچs‚¤ڈêچ‡‚ة‚حپACRISPR Cas9/sgRNA ‚ـ‚½‚ح TALEN ƒxƒNƒ^پ[‚ج‚ف‚ًˆê‰كگ«“±“üپiƒvƒ‰ƒXƒ~ƒhƒgƒ‰ƒ“ƒXƒtƒFƒNƒVƒ‡ƒ“پj‚µ‚ـ‚·پBژہچغ‚ةچ×–EƒQƒmƒ€‚ً•زڈW‚·‚éچغ‚ة‘ٹ“¯‘gٹ·‚¦—p‚جƒhƒiپ[ƒxƒNƒ^پ[‚ھ•K—v‚¾‚ئ‚µ‚ؤ‚àپA‚±‚جŒںڈطژہŒ±‚ة‚حƒhƒiپ[ƒxƒNƒ^پ[‚ح•s—v‚إ‚·پBIndelCheck™ ƒVƒXƒeƒ€‚إ‚حپAƒhƒiپ[ƒxƒNƒ^پ[‚ج—L–³‚ةٹض‚ي‚炸پANHEJ ‚ة‚و‚蓱“ü‚³‚ꂽ•دˆظ‚ًŒںڈo‚µ‚ـ‚·پB
ژہŒ±ژèڈ‡‚ئ‚µ‚ؤ‚حپA‚ـ‚¸پAƒgƒ‰ƒ“ƒXƒtƒFƒNƒVƒ‡ƒ“‚µ‚ؤ1-2“ْŒمپAچ×–Eƒvپ[ƒ‹‚ً‰ٌژû‚µ‚ؤپAƒ^پ[ƒQƒbƒg•”ˆت‹ك–T‚ةگفŒv‚µ‚½ƒvƒ‰ƒCƒ}پ[‚ً—p‚¢‚ؤPCR‚ًچs‚¢‚ـ‚·پB

گ}2. IndelCheck™ ƒVƒXƒeƒ€—pPCRƒvƒ‰ƒCƒ}پ[‚جگفŒv•û–@
‰©گF‚جƒnƒCƒ‰ƒCƒg‚إژ¦‚·”z—ٌ‚ًPCRƒvƒ‰ƒCƒ}پ[Œ‹چ‡•”ˆت‚ئ‚µ‚½پBپiPCRژY•¨‚جگ„ڈ§ƒTƒCƒY: 500-1,000 bpپjPCRƒvƒ‰ƒCƒ}پ[‚ًگفŒv‚·‚éچغپA“ٌ–{چ½گط’fˆت’u‚حƒvƒ‰ƒCƒ}پ[گفŒv—جˆو‚ج’†ٹش“_‚©‚çڈ‚ب‚‚ئ‚à 100 bp “à‚ة‚ب‚é‚و‚¤گفŒv‚·‚é‚و‚¤’چˆس‚·‚éپBƒ^پ[ƒQƒbƒg•”ˆت‚ح3‚©ڈٹ‚ئ‚µپi—خگFپAƒsƒ“ƒNگFپAگ…گFپjپA‚¤‚ـ‚ƒ~ƒXƒ}ƒbƒ`‚ھŒںڈo‚³‚ê‚éڈêچ‡پA‰؛‹L‚جƒTƒCƒY‚جƒoƒ“ƒh‚ھŒںڈo‚³‚ê‚éپB
human HDAC6ˆâ“`ژq‚جPCRژY•¨پF989 bpپiNCBI GeneID: 9636پj
341 bp + 648 bpپi—خگFپj
555 bp + 434 bpپiƒsƒ“ƒNگFپj
616 bp + 373 bpپiگ…گFپj
NHEJ ‚ً‰î‚µ‚½•دˆظ“±“ü‚حٹm—¦“I‚إپA‘}“üپAŒ‡ژ¸‚ا‚؟‚ç‚à‹N‚±‚蓾‚ـ‚·پB‚ـ‚½پA‘}“üŒ‡ژ¸”z—ٌ‚ج’·‚³‚حژہŒ±–ˆ‚ةˆظ‚ب‚è‚ـ‚·پBNHEJ ‚ً‰î‚µ‚½•دˆظ“±“ü‚ھگ¬Œ÷‚µ‚½ڈêچ‡پAچ×–Eƒvپ[ƒ‹‚ة‚حƒ^پ[ƒQƒbƒg•”ˆت‚ةˆظ‚ب‚é•دˆظ‚ً‚à‚آ—lپX‚بڈW’c‚ھٹـ‚ـ‚ê‚ـ‚·پB‚ـ‚½پAچ×–Eƒvپ[ƒ‹‚ة‚ح•زڈW‚³‚ê‚ؤ‚¢‚ب‚¢–ىگ¶Œ^‚جڈW’c‚àٹـ‚ـ‚ê‚ـ‚·‚ج‚إپA—lپX‚بچ×–EڈW’c‚ھچ¬چف‚µ‚ؤ‚¢‚éڈَ‘ش‚ح”ٌڈي‚ة•،ژG‚¾‚ئچl‚¦‚ç‚ê‚ـ‚·پB
PCR‚إ‚ج‘•ŒمپAژY•¨‚ً 95پژ ‚إ•دگ«‚µپAڈي‰·‚ة‚ب‚é‚ـ‚إ‚ن‚ء‚‚è‚ئ—â‚ـ‚·‚±‚ئ‚إچؤƒAƒjپ[ƒ‹‚µ‚ـ‚·پB—â‹p‚ة‚و‚èپAƒ}ƒbƒ`”z—ٌپAƒ~ƒXƒ}ƒbƒ`”z—ٌ‚ة‚آ‚¢‚ؤ‚»‚ꂼ‚êƒAƒjپ[ƒٹƒ“ƒO‚ھ‹N‚±‚èپAƒzƒ‚“ٌ–{چ½‚¨‚و‚رƒwƒeƒچ“ٌ–{چ½‚ھگ¶‚¶‚ـ‚·پiگ}1پjپB
‚»‚جŒمپAچؤƒAƒjپ[ƒٹƒ“ƒO‚µ‚½DNA‚ً T7 Endonuclease I ‚إڈˆ—‚µ‚ـ‚·پBT7 Endonuclease I ‚ح“ٌ–{چ½‚جƒ~ƒXƒ}ƒbƒ`‚ً”Fژ¯‚µپAƒwƒeƒچ“ٌ–{چ½‚جƒ~ƒXƒ}ƒbƒ`•”•ھ‚ًگط’f‚µ‚ـ‚·‚ھپAƒzƒ‚“ٌ–{چ½‚حگط’f‚³‚ê‚ـ‚¹‚ٌپBT7 Endonuclease I ‚إڈء‰»‚µ‚½’f•ذ‚حƒAƒKƒچپ[ƒXƒQƒ‹‚إ“d‹C‰j“®‚µ‚ـ‚·پBˆê”ت“I‚ةپANHEJ ‚ً‰î‚µ‚½•دˆظ‚ھ“±“ü‚³‚ê‚é‚ئ3‚آ‚جƒoƒ“ƒh‚ھŒ©‚ç‚ê‚ـ‚·پB1‚آ–ع‚حگط’f‚³‚ê‚ب‚©‚ء‚½DNAپiƒzƒ‚“ٌ–{چ½پjپA‘¼2–{‚جƒoƒ“ƒh‚حگط’f‚³‚ꂽ’f•ذپiƒwƒeƒچ“ٌ–{چ½پj‚إ‚·پiگ}3پjپB
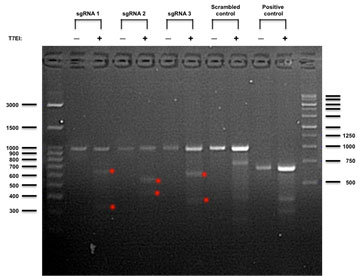
گ}3. ƒ~ƒXƒ}ƒbƒ`گط’f’f•ذ‚ج“d‹C‰j“®Œ‹‰ت
HEK293 چ×–E‚ة CRISPR/sgRNA ƒNƒچپ[ƒ“‚ًƒgƒ‰ƒ“ƒXƒtƒFƒNƒVƒ‡ƒ“‚µپAIndelCheck™ ƒVƒXƒeƒ€‚ة‚ؤ•دˆظŒںڈo‚ًچs‚ء‚½پBƒAƒjپ[ƒٹƒ“ƒO‚µ‚½DNA’f•ذ‚ً T7 Endonuclease I ‚إڈء‰»‚µ‚½ƒTƒ“ƒvƒ‹پiپ{پjپAڈء‰»‚µ‚ؤ‚¢‚ب‚¢ƒTƒ“ƒvƒ‹پi-پj‚ة‚آ‚¢‚ؤ 2% ƒAƒKƒچپ[ƒXƒQƒ‹‚إ•ھ—£‚µ‚½پB
پ–ˆَ‚إژ¦‚µ‚½ƒ~ƒXƒ}ƒbƒ`گط’f’f•ذ‚ھŒںڈo‚³‚ꂽƒTƒ“ƒvƒ‹‚ة‚آ‚¢‚ؤ‚ح CRISPR ‚ھچ×–E“à‚إ‹@”\‚µ‚½‚ئچl‚¦‚ç‚ê‚éپB’تڈيپAƒXƒNƒ‰ƒ“ƒuƒ‹ sgRNA ƒRƒ“ƒgƒچپ[ƒ‹‚إ‚ح“ٌ–{چ½گط’f‚ح‹N‚±‚ç‚ب‚¢‚ھپA–{ژہŒ±‚إ‚حپAƒXƒNƒ‰ƒ“ƒuƒ‹ sgRNA ƒRƒ“ƒgƒچپ[ƒ‹ŒQ‚إ T7 Endonuclease I ڈء‰»Œم‚ةƒ~ƒXƒ}ƒbƒ`گط’f’f•ذ‚ھŒںڈo‚³‚ꂽپB‚±‚ê‚حپAژ©‘R‚ة‘¶چف‚·‚éˆظ‚ب‚éگُگF‘جڈم‚جˆâ“`ژq‘½Œ^‚ة‚و‚é‚à‚ج‚ئچl‚¦‚ç‚ê‚éپBƒXƒNƒ‰ƒ“ƒuƒ‹ƒRƒ“ƒgƒچپ[ƒ‹ŒQ‚جگط’f’f•ذ‚ج‘ه‚«‚³‚ح sgRNA ŒQپi1, 2, 3پj‚جگط’f’f•ذ‚ئ‚حˆظ‚ب‚ء‚ؤ‚¢‚邽‚كپA‹^—zگ«‚إ‚ح‚ب‚¢‚ئچl‚¦‚ç‚ê‚éپB
HEK293 چ×–E‚ة CRISPR/sgRNA ƒNƒچپ[ƒ“‚ًƒgƒ‰ƒ“ƒXƒtƒFƒNƒVƒ‡ƒ“‚µپAIndelCheck™ ƒVƒXƒeƒ€‚ة‚ؤ•دˆظŒںڈo‚ًچs‚ء‚½پBƒAƒjپ[ƒٹƒ“ƒO‚µ‚½DNA’f•ذ‚ً T7 Endonuclease I ‚إڈء‰»‚µ‚½ƒTƒ“ƒvƒ‹پiپ{پjپAڈء‰»‚µ‚ؤ‚¢‚ب‚¢ƒTƒ“ƒvƒ‹پi-پj‚ة‚آ‚¢‚ؤ 2% ƒAƒKƒچپ[ƒXƒQƒ‹‚إ•ھ—£‚µ‚½پB
پ–ˆَ‚إژ¦‚µ‚½ƒ~ƒXƒ}ƒbƒ`گط’f’f•ذ‚ھŒںڈo‚³‚ꂽƒTƒ“ƒvƒ‹‚ة‚آ‚¢‚ؤ‚ح CRISPR ‚ھچ×–E“à‚إ‹@”\‚µ‚½‚ئچl‚¦‚ç‚ê‚éپB’تڈيپAƒXƒNƒ‰ƒ“ƒuƒ‹ sgRNA ƒRƒ“ƒgƒچپ[ƒ‹‚إ‚ح“ٌ–{چ½گط’f‚ح‹N‚±‚ç‚ب‚¢‚ھپA–{ژہŒ±‚إ‚حپAƒXƒNƒ‰ƒ“ƒuƒ‹ sgRNA ƒRƒ“ƒgƒچپ[ƒ‹ŒQ‚إ T7 Endonuclease I ڈء‰»Œم‚ةƒ~ƒXƒ}ƒbƒ`گط’f’f•ذ‚ھŒںڈo‚³‚ꂽپB‚±‚ê‚حپAژ©‘R‚ة‘¶چف‚·‚éˆظ‚ب‚éگُگF‘جڈم‚جˆâ“`ژq‘½Œ^‚ة‚و‚é‚à‚ج‚ئچl‚¦‚ç‚ê‚éپBƒXƒNƒ‰ƒ“ƒuƒ‹ƒRƒ“ƒgƒچپ[ƒ‹ŒQ‚جگط’f’f•ذ‚ج‘ه‚«‚³‚ح sgRNA ŒQپi1, 2, 3پj‚جگط’f’f•ذ‚ئ‚حˆظ‚ب‚ء‚ؤ‚¢‚邽‚كپA‹^—zگ«‚إ‚ح‚ب‚¢‚ئچl‚¦‚ç‚ê‚éپB
IndelCheck™ ƒVƒXƒeƒ€‚ً—p‚¢‚½ƒXƒNƒٹپ[ƒjƒ“ƒOژہŒ±
IndelCheck™ ƒVƒXƒeƒ€‚حپANHEJ ‚ً‰î‚µ‚½ƒmƒbƒNƒAƒEƒg‚جƒXƒNƒٹپ[ƒjƒ“ƒO‚ة‚àژg—p‰آ”\‚إ‚·پBƒXƒNƒٹپ[ƒjƒ“ƒO‚جژèڈ‡‚حڈمڈq‚جŒںڈط•û–@‚ئ“¯—l‚إ‚·‚ھپAچ×–EƒTƒ“ƒvƒ‹‚حˆظ‚ب‚éƒAƒŒƒ‹‚ًٹـ‚قچ×–EڈW’c‚جƒ~ƒbƒNƒX‚إ‚ح‚ب‚’PˆêƒNƒچپ[ƒ“‚إ‚ ‚é•K—v‚ھ‚ ‚è‚ـ‚·پB’PˆêƒNƒچپ[ƒ“‚ھ“¯ˆê‚ج•دˆظ‚ًƒzƒ‚‚إ‚à‚ء‚ؤ‚¢‚éڈêچ‡پAIndelCheck™ ƒVƒXƒeƒ€‚إ‚حƒwƒeƒچ“ٌ–{چ½DNA‚حگ¶‚¶‚¸پA“d‹C‰j“®ƒfپ[ƒ^‚©‚ç‚حپA‚»‚جƒNƒچپ[ƒ“‚ھ•دˆظ“±“üچ×–E‚إ‚ ‚é‚ئ‚ح”»’f‚³‚ê‚ـ‚¹‚ٌپB‚±‚ج‚و‚¤‚بڈَ‹µ‚ً”ً‚¯‚邽‚كپAT7 Endonuclease I ڈˆ—‚ًچs‚¤‘O‚ةپA–ىگ¶Œ^‚جPCRژY•¨‚ًƒXƒpƒCƒNƒCƒ“‚·‚邱‚ئ‚ً‚¨ٹ©‚ك‚µ‚ـ‚·پB
‚±‚جƒXƒeƒbƒv‚ة‚و‚èƒwƒeƒچ“ٌ–{چ½DNA‚ًŒںڈo‚µپA•دˆظ‚ج—L–³‚ًŒ©•ھ‚¯‚邱‚ئ‚ھ‰آ”\‚إ‚·پB•دˆظ‚ھ“±“ü‚³‚ꂽƒNƒچپ[ƒ“‚ھ“¯’肳‚ꂽ‚çپA•دˆظ‚¨‚و‚رƒAƒŒƒ‹‚ً’²‚ׂ邽‚كپATAƒTƒuƒNƒچپ[ƒjƒ“ƒO‚âƒVپ[ƒNƒGƒ“ƒX‰ًگح‚ًچs‚¢‚ـ‚·پB

ڈ¤•i‚حپuŒ¤‹†—pژژ–ٍپv‚إ‚·پBگl‚â“®•¨‚جˆم—أ—pپE—صڈ°گf’f—pپEگH•i—p‚ئ‚µ‚ؤ‚ح
ژg—p‚µ‚ب‚¢‚و‚¤‚ةپAڈ\•ھ‚²’چˆس‚‚¾‚³‚¢پB