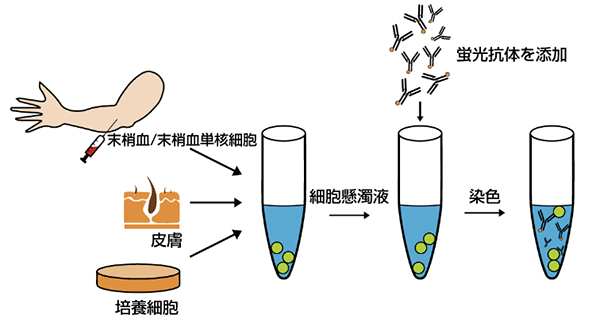
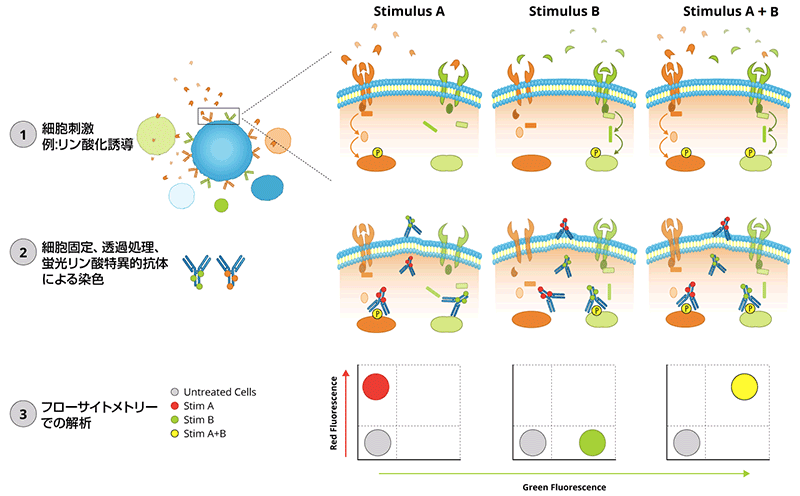
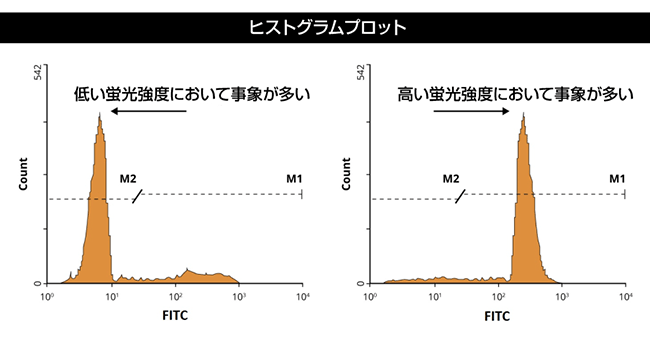
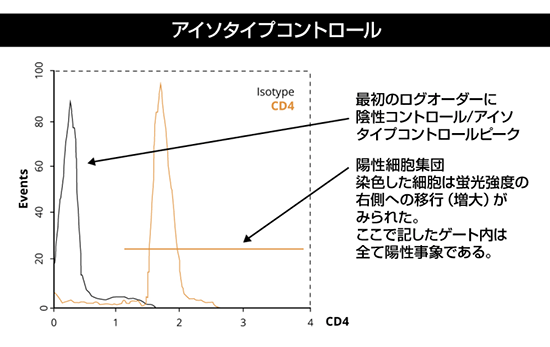
ここでは、フローサイトメトリー実験の段階ごとにその手順の概説をします。本情報は、あくまで一般的なガイドであり、実際にフローサイトメトリーの実験を行う際には使用する細胞種に応じてプロトコールを最適化する必要があります。
一般的なFACS染色プロトコール
細胞は数百〜数千もの細胞表面抗原を発現し、これが細胞種、生物学的機能、発生段階などを特定する材料となります。異なる臓器に存在する細胞は、それぞれ特徴的な細胞表面抗原をもつため、この抗原に特異的な蛍光標識抗体を用いたフローサイトメトリー解析により細胞の識別が可能です。非標識抗体を用いる場合には、標識二次抗体による染色ステップが追加で必要となります(非直接的免疫染色)。
注:もし細胞を96ウェルのU底あるいはV底プレートで染色する場合、非結合一次抗体を最大限に除去するための洗浄ステップが必要です。
1 表面抗原の直接蛍光免疫染色
主な試薬 : PBS、染色緩衝液、FACS緩衝液、PFA固定緩衝液
- 適切なプロトコールに準じて単一細胞懸濁液を作製し、染色緩衝液で細胞濃度を107 cells/mlに調製する。
- 100µLの細胞懸濁液を必要数(未染色コントロール、補正コントロール、オプションでイソ型やFMOコントロール、および試験サンプル)の染色チューブに入れる。
- 至適希釈した抗体を対応するチューブに入れ、4°C(氷上)で30分間、暗所で培養する。
- 氷冷PBSを細胞に添加して洗浄し、300〜400xgで遠心した後、100〜200µLのFACS緩衝液/PFA固定緩衝液に再懸濁する。
- 4°C, 暗所で保管し、24時間以内にデータ取得することが望ましい。
2 表面抗原の間接蛍光免疫染色
主な試薬: PBS、染色緩衝液、FACS緩衝液、PFA固定緩衝液
- 適切なプロトコールを用いて単一細胞懸濁液を調製し、染色緩衝液を用いて細胞濃度を107 cells/mlに調製する。
- 100µLの細胞懸濁液を必要数(未染色コントロール、補正コントロール、オプションでイソ型やFMOコントロール、および試験サンプル)の染色チューブに入れる。
- 至適希釈した抗体を対応するチューブに入れ、4°C(氷上)で30分間、培養する。
- 氷冷PBSを細胞に添加して洗浄し、300〜400xgで遠心した後、100µLの染色緩衝液に再懸濁する。
- 適切な希釈率の特定二次抗体を添加し、細胞を4°C(氷上)で30分間、暗所で培養する。
- 氷冷PBSを細胞に添加して洗浄し、300〜400xgで遠心した後、100〜200µLのFACS緩衝液/PFA固定緩衝液に再懸濁する。
- 4°Cの暗所で保存し、24時間以内にデータ取得することが望ましい。
3 細胞内抗原の一般的な免疫染色手順
A. メタノールによる透過処理
主な試薬 : PBS、染色緩衝液、FACS緩衝液、0.5 〜 4% PFA含有PBS(厳密なPFA濃度を基準として全ての抗体パネルに使用)、100%メタノール
- 適切なコントロールと共に、プロトコール1または2に準じて表面染色を行う。
- 染色済み細胞を107 cells/mlとなるよう0.5〜4% PFAに分注する。未染色の分注品を細胞内染色コントロールとして調製する。
- 細胞を10〜30分間、遮光して固定する。
- 300〜400 xgで遠心して固定液を洗い落とし、氷冷した100%メタノールをゆっくり添加してゆっくりとボルテックスする。
- 細胞を氷上で30分間、遮光して培養する。
- 400〜500 xgで遠心してメタノールを洗い落とし、細胞を100µLの染色緩衝液に再懸濁する。
- 100µLの細胞懸濁液を必要数の染色チューブ(未染色コントロール、補正コントロール、オプションでイソ型やFMOコントロール、および試験サンプル)に入れる。
- 至適条件で希釈した一次抗体を対応するチューブに入れ、4°C(氷上)で30分間、培養する。
- 氷冷PBSを細胞に添加し、400〜500 xgで遠心した後、100µLの染色緩衝液に再懸濁する。
- 適切な希釈濃度の二次抗体を添加し、細胞を4°C(氷上)で30分間、暗所で保存する。
- 細胞を冷却PBSで洗浄し、400〜500 xgで遠心後、100〜200µLのFACS緩衝液/PFA固定緩衝液に再懸濁する。
- 遮光して4°Cで保存し、24時間以内にデータ取得することが望ましい。
4 細胞内サイトカイン / リン酸免疫染色
細胞内染色ステップでは、表面抗原決定だけでなく、同時に細胞質内や核に存在する抗原(サイトカインや転写因子)の直接測定が可能となります。このステップでは、蛍光標識した表面抗原と染色後に細胞の固定そして透過処理が必要です。血液や他の組織由来のあらゆる細胞の機能活性を、特別な分離処理をすることなく、直接測定することができます。よりよい結果を得るためには、細胞内でサイトカイン産生を増大させるために分裂促進因子(PMA, Ca++、またはペプチド抗原決定基やタンパク質輸送阻害剤、ブレフェルジンAなど)によるin vitro細胞刺激が必要な場合もあります。
主な試薬 : PBS、染色緩衝液、FACS緩衝液、0.5%PFA含有PBS、0.1%サポニンまたは100%メタノール、適切な細胞刺激剤、ブレフェルジンAまたはモネンジン
注:もし細胞を96ウェルのU底あるいはV底プレートで染色する場合、非結合一次抗体を最大限に除去するための洗浄ステップが必要です。
- 適切なプロトコールに準じて細胞を回収し、予め決定した濃度で細胞をチューブに分注する(細胞種や刺激剤による)。
- 刺激剤を添加し、細胞を37°Cで必要時間培養する。次項の表は、標的タンパク質に対する刺激剤と培養時間の参照である。
- ・分泌サイトカインを染色する場合、ブレフェルジンAまたはモネンジンを製造元の推奨濃度で培養期間中に添加する。
- ・未染色コントロールや染色基線コントロールとして未刺激の分注物を取り置く。
- 最終濃度が0.5% PFAで細胞を固定し、刺激を止める。
- 緩やかにボルテックスし、細胞を氷上に15分間静置する。
- 固定液を洗い落とし、プロトコール1または2に準じて表面染色を行う。
- 適切な透過処理試薬に細胞を再懸濁し、プロトコール3aまたは3bに準じて透過処理と細胞内染色を実施する(100%メタノールまたは0.1%サポニン)。
5 In Vitro 細胞刺激剤参照表
| 標的サイトカイン/リン酸化タンパク質 |
標的細胞 |
刺激物 |
処理時間 |
表面マーカー |
| IL-2 |
PBMC |
PMA (50ng/ml) |
4-6 hours |
CD3 |
| IL-3 |
T-細胞 |
PMA(50ng/ml) + ionomycin (1µg(ml) |
4-6 hours |
CD4 |
| IL-4 |
PBMC |
PMA(50ng/ml) + ionomycin (1µg(ml) |
4-6 hours |
CD4 |
| IL-6 |
PBMC |
LPS (100ng/ml) |
4-6 hours |
CD14 |
| IL-10 |
PBMC |
LPS (100ng/ml) |
18-24 hours |
CD14 |
| GM-CSF /IFNγ/TNFα/TNFβ |
PBMC |
PMA(50ng/ml) + ionomycin (1µg(ml) |
4-6 hours |
CD3 |
| pStat5 |
PBMC |
GM-CSF (20ng/ml) + IL3 (20ng/ml) |
15 min. |
CD123, CD116 |
| pStat3 |
PBMC |
G-CSF (20ng/ml) + IL6 (20ng/ml) |
15 min. |
CD126, CD114 |
| pERK |
PBMC |
IL3 (20ng/ml) + IL6 (20ng/ml) + FLT3L (20ng/ml) |
15 min. |
CD123, CD126, CD135 |
6 Dye efflux staining (分染法)
色素除外染色(分染法)は、生細胞と死細胞を分離するとともに、希少な幹細胞“サイドポピュレーション”の単離に使用されています。免疫染色の際に生死判定の染色を日常的に行っている場合、他の染色を行う前に実施したほうがよいでしょう。
A. ヨウ化プロピジウム(PI)染色(生存率)
主な試薬 : PBS、染色緩衝液、PI溶液(10µg/mlのPBS溶液)
- 細胞を回収し、PBSで1回洗浄する。
- 細胞を染色緩衝液に107 cells/mLで再懸濁する。
- 100µlの細胞懸濁液に対して5µlのPI染色液を添加し、緩やかに混合して遮光して1分間静置する。
- 染料を洗い落とし、細胞を適切な容量の染色緩衝液に再懸濁する。
B. 7-アミノアクチノマイシン D (7-AAD) 染色 (生存率)
主な試薬 : PBS、染色緩衝液、7-ADD溶液(100µg/mlのPBS溶液)
- 細胞を回収し、PBSで1回洗浄する。
- 細胞を染色緩衝液に107 cells/mLで再懸濁する。
- 100µlの細胞懸濁液に対して2µlの7-ADD染色液を添加し、緩やかに混合して30分間氷上で静置する。
- 染料を洗い落とし、細胞を適切な容量の染色緩衝液に再懸濁する。
C. ローダミン 123 および Hoechst 33342染色(サイドポピュレーション)
主な試薬 : PBS、5% FBSのPBS溶液、染色緩衝液、Hoechst 33342溶液(1mMのPBS溶液)またはローダミン123溶液(10µg/mlのPBS溶液)
- 細胞を回収し、PBSで1回洗浄する。
- 細胞を5% FBSに107 cells/mLで再懸濁する。
- 100µlの細胞懸濁液に対して1µlの染色液を添加し、37°Cのウォーターバスで30分間インキュベートする。
- 染料を洗いおとし、細胞を予め温めた色素を含まない5% FBSに再懸濁した後、37°Cのウォーターバスで30分間インキュベートする。
- 細胞を再度洗浄し、適切な容量の染色緩衝液に再懸濁する。
7 DNA含有量または細胞周期解析
主な試薬 : PBS、染色緩衝液、PI溶液(50µg/mlのPBS溶液)、RNase A(10µg/ml)、70%エタノール
- 細胞を回収し、PBSで1回洗浄する。
- 細胞を染色緩衝液に107 cells/mLで再懸濁する。
- 500µlの細胞を予め冷却した別のチューブに分注し、氷冷した70%エタノールを滴下しながら緩やかにボルテックスする。
- 細胞を氷上に1時間静置する。
- 細胞にPBSで洗浄し、400〜500 xgで10分間遠心する。2回繰り返す。
- 1mLのPI溶液を細胞沈殿物に添加し、よく混合する。50µLのRNaseを最終濃度0.5µg/mLとなるように添加する。
- 細胞を4°Cで一晩培養する。
- 細胞をPBSで1回洗浄後、適切な容量の染色緩衝液に再懸濁する。
全てのフローサイトメトリー(またはFACS)実験は、サンプル調製から始まります。重要なことは、機器の詰まりを防ぎ、高品質かつ一貫した結果を得るために解析する細胞を単一細胞懸濁液とすることです。以下のサンプル調製ガイドをご参照いただき、フローサイトメトリー解析用のサンプル調製方法をご確認ください。
染色用の細胞調製
フローサイトメトリー解析用の細胞は、おもに4種類の供給源に由来します。
- 接着または浮遊培養
- 凍結保存サンプル
- 全血またはバフィーコート
- 固体組織:例えば、骨髄、脾臓、腸など
- 凝集塊や壊死組織片のなり均一な単一細胞懸濁液
- 細胞密度106-107 細胞/mL
- 適切な染色緩衝液に懸濁
全血からフィコール密度勾配遠心分離により単離したPBMCや赤血球可溶化全血、非接着培養細胞は、フローサイトメトリー解析にそのまま使用することができます。接着培養細胞や固体臓器に存在する細胞は、フロー解析を行う前にそれぞれ酵素処理または組織の物理的解離を行い、まず単一細胞懸濁液にする必要があります。その後、機器の詰まりやフローデータの質の低下を避けるため、フィルターでろ過します。使用するサンプルに応じて、以下のサンプル調製プロトコールをご検討ください。
◆ 浮遊培養細胞の調製
主な試薬 : PBS、染色緩衝液
- 細胞を、組織培養容器から遠心チューブへと流し入れる。
- 300 - 400 xgで5〜10分間、室温で遠心する。
- 上清を廃棄し、沈殿物をPBSに再懸濁し、上記ステップを再度行う。
- 上清を廃棄し、適切な容量の冷却済み染色緩衝液に再懸濁する。
◆ 接着培養細胞の調製
主な試薬 : PBS, 染色緩衝液、0.25%トリプシン
- 培養培地を廃棄し、一層の細胞を滅菌PBSで洗浄する。
- 温めたトリプシンを一層の細胞がちょうど覆われる程度に添加し、37°で5〜10分間(細胞種による)培養する。
- 培養培地(血清添加培地)で反応を中和し、緩やかに容器を震盪させて細胞を剥がす。
- 以降、浮遊培養細胞調製プロトコールに準ずる。
◆ 凍結保存細胞の調製
主な試薬 : PBS, 染色緩衝液、10% FBS含有培養培地
- クリオチューブを直ちに37°Cの温浴に入れ、融解する。
- 冷却した遠心チューブに移し、氷冷した培養培地を滴下して細胞が10x希釈になるようにする。氷上操作すること。
- 300〜400xgで5分間、4°Cで遠心する。
- 上清を廃棄し、冷却した染色緩衝液で1回洗浄する。
- 適切な容量の冷却した染色緩衝液で細胞を再懸濁する
◆ 全血またはバフィーコートからの細胞調製
主な試薬 : PBS, 染色緩衝液、フィコールやヒストパックなどの適切な密度勾配培地
- 全血またはバフィーコートを室温に戻した等量のPBSで希釈する。
- 等量の密度勾配培地上に希釈全血を注意して層状にのせる。
- 400〜500 xgで30〜40分間、室温で遠心する。遠心機のブレーキはオフにしておく。
- 上相の血漿層と下相の培地相の間の薄い境界面よりPBMCを吸引する。
- 顆粒球を除去するため、RBC沈降物のすぐ上の白っぽい色の相を吸引する。
注:異なる顆粒球集団を濃縮するための特殊化された密度勾配培地が処方されている。
- 細胞をPBSに再懸濁し、300〜400 xgで10分間、室温で遠心する。
- PBSでさらに1回あるいは2回洗浄する。
- 細胞を適切な容量の染色緩衝液に再懸濁する。
◆ マウス骨髄からの細胞調製
主な試薬 : PBS、染色緩衝液(付録参照)、RBS可溶化緩衝液(処方は付録参照、または市販の緩衝液を使用)
- 頸骨や大腿骨を取り出し、無関係な脂肪、筋肉、および結合組織を全て除去する。骨を冷却したPBSに入れ、氷上保管する。以降の操作は全て氷上で行う。
- シリンジを冷却した培養培地で満たし、18ゲージニードルを取り付ける。骨の端に針で穴を開け、組織培養用処理済ではないプレート上に中身を流し出す。
- 22ゲージニードルを用いて細胞の塊を単一細胞懸濁液へと分離する。
- 細胞を300〜400 xgで遠心沈降させ、さらに1回冷却PBSで洗浄する。
- 細胞を適切な容量の染色緩衝液に再懸濁させ、細胞を再度沈降させる前に細胞数をカウントする。
- 細胞を106 cells/mlとなるようRBC可溶化緩衝液に再懸濁し、室温で5分間培養する。
- 細胞を遠心沈降させ、可溶化緩衝液を廃棄後、PBSで1回洗浄する。
- 細胞を適切な容量の染色緩衝液で再懸濁する。
本稿では一般的なフローサイトメトリーの実験手順をご説明します。参照ガイドとしてご利用いただき、詳細はご利用になる商品のデータシートをご確認ください。
フローサイトメトリー実験の最適化
フロー手順のほぼ全てのステップで、選択肢があります。どの細胞固定法や透過処理方法が最適なのか。多色染色にはどの蛍光染色を使うべきか。使用する抗体にはどのイソ型コントロールが適切なのか。ここでは、フローサイトメトリー実験における最適化のポイントをご紹介します。
◆ サンプル調製最適化
高品質なフローデータを得るための最初のステップはサンプル調製です。単一細胞懸濁液を調製する非常に重要なステップであり、予期せぬ装置の詰まりや低品質なデータを避けることができます。
- 可能な限り、凍結融解した細胞ではなく、新しく単離した細胞を使用する。
- 融解細胞の生存率を上げるため、融解細胞の初期希釈を高血清濃度下(90%FBSの培養培地)で行う。
- 接着細胞が多い集団を単離する場合は、組織培養非処理のディッシュやチューブを使用する。
- 複雑な組織から細胞を単離する場合、食作用や細胞融解を防ぐため氷上操作する方がよい(消化酵素を使用するステップは除く)。
- 均一な単一細胞懸濁液を調製し、細胞崩壊を防ぐため、ボルテックスにかけるのではなく、できるだけ優しくピペッティングを行う。
- もし、下流のステップで生細胞選別を行う場合、各ステップで細胞を計数して生存率を確認する。
- ナイロンメッシュを通して細胞塊や他の破片を除去し、最終懸濁液中の死細胞数を最小限に抑える。
◆ 透過処理と固定法の最適化
抗体を細胞内のタンパク質に期待通りにバインドさせるためのさまざまな透過処理や固定法があります。細胞固定により標的タンパク質を元来の細胞内部位に維持することができ、可溶性抗原や半減期の短い抗原の安定性が向上します。
- サイトカインのような分泌タンパク質の染色には、サイトカインを内部に閉じ込めるため、固定/透過処理の前にモネンジンやブレフェルジンAといったタンパク質輸送阻害剤を添加する。
- 表面と細胞内の染色を組み合わせて行う場合、固定/透過処理で抗原決定基を変更し抗体結合に影響を及ぼす場合があるため、まず表面マーカーを染色した後、固定/透過処理を行う方がよい。
- 固定/透過処理試薬は細胞の散乱特性や自己蛍光を変化させる。したがって、同一試薬で処理した未染色コントロールを含むことを推奨する。
- 抗体の表面抗原への結合で細胞を刺激し、細胞内シグナルタンパク質の発現に影響する場合がある。したがって、表面染色は刺激後に実施するべきである。
- リン酸化は一時的であるため、フォスフロー染色では、細胞を刺激後、直ちに固定して透過処理する。
- 適切な透過処理剤の選択が非常に重要である。サポニンまたはトリトンXやメタノールのようなデタージェントを選択することもある。
- サポニンは表面抗原決定基を変更しないため、その後で表面染色を行うことができる。
- トリトンXやTweenは長期培養により細胞を可溶化するため、避けた方がよい。
- メタノールはほとんどの細胞内抗原と互換性があり、メタノール処理した細胞はシグナルを失うことなく、-20〜-80°Cで長期保存できる。
- 蛍光色素によってはメタノール処理に耐えられないものもある。以下の表に一般的に使用される色素をメタノール耐性と感受性にわけて示した。
| メタノール感受性 |
メタノール耐性 |
| FITC |
PE |
| eFlour 450 |
APC |
| eFluor 660 |
|
| Alexa Fluor 647 |
|
| Alexa Fluor 488 |
|
| PerCP |
|
| 全てのタンデム型色素 |
|
フロー実験は、複数のパラメーターや混合細胞集団、時には数々の実験コントロールが関与するなど、非常に複雑になる場合があります。ここでは、FACS実験に適切なコントロールを選択するためのガイドをご紹介します。
FACSコントロールの選択
フローサイトメトリー実験計画においてはじめに考慮するべきことのひとつとして、コントロールの選択が挙げられます。実験における誤差や生存率にはさまざまな原因があり、これらを制御することが非常に重要です。コントロールの目的は、フローサイトメトリー機器が適切に機能していること、正確に設定されていること、適切なゲートが描かれ、データの正しい解釈が得られることを担保することです。どのコントロールが目的のフローサイトメトリー実験に適切かを判断する指標として、以下の表をご参照ください。
| コントロール |
何を加えるか |
目的 |
注釈 |
| 未染色コントロール |
未染色細胞(染色サンプルと並行して培養)に抗体を添加せず、そのまま最後まで処置。 |
自家蛍光(発光)由来のバックグランドを制御し、電圧や陰性ゲートを適切に設定するため。追加の陰性コントロールとして利用。 |
ビーズとの比較により相対自家蛍光(発光)の決定に役立つ。自家蛍光(発光)レベルが高い場合、異なる励起源を試行する。 |
| イソ型コントロール |
イソ型コントロール抗体と共に培養した細胞(一般に、対象細胞に存在しないはずの抗原に対して産生された抗体) |
一次抗体の非特異的結合を測定するため。 |
イゾ型コントロールは以下の事項と一致すべきである。
- 宿主生物種
- 抗体のIgサブクラス(IgA, IgG, IgD, IgE, or IgM)
- 一次抗体と同一の蛍光標識
|
| 内部陰性コントロール |
対象抗原を発現しない細胞集団であり、かつ最後まで処置したもの。 |
非特異的抗体結合による偽陽性を防ぐため。 |
陰性コントロール細胞がいつも存在するとは限らない。理想的には、内部コントロールの蛍光強度は未染色コントロールのものと同等である。 |
| 陽性コントロール |
実験対象を発現することが既知の細胞。 |
質の悪い抗体による偽陰性を避けるため。 |
陽性コントロール細胞が必ず存在するとは限らない。 |
| 補正コントロール |
補正コントロールは的確な実験蛍光色素(類似した輝度をもつもの)と一致しなくてはならない。コントロールは、補正対象サンプル全てと少なくとも同等かそれ以上の明るさが必要であり、バックグラウンドの蛍光は陽性と陰性コントロールで同等であるべきである。 |
スペクトルの重複を除去(補正)することが可能となる。スペクトルの重複は使用する全ての蛍光色素に対して補正する。 |
異なる蛍光色素間でのスペクトル重複のため、多色フローサイトメトリーでの単一染色は重要である。統計学的に有意となるよう十分な事象を収集する。 |
| 生存率コントロール |
死細胞を同定する一般的な生存能力色素には、DNA色素やタンパク質結合色素がある。 |
生存率染色による死細胞除外することで、非特異的結合の低減と陽性染色集団の同定が容易となる。 |
生/死染色を利用して死細胞を除去することで、染色が改善する。死細胞はより大きな自己蛍光発光をもち、非特異的抗体結合を増大させるため、偽陽性やダイナミックレンジの減少の原因となる。 |
| Fc 遮蔽コントロール |
染色手順においてFc 遮断試薬を添加。市販のFc遮断薬を利用できる。 |
抗原特異的結合のみが生ずることを保証するため(Fc受容体を介した抗体結合が偽陽性を誘導し、データの解釈が不能となる)。 |
代替法として、宿主血清の一次抗体を使用することもできる。例えば、対象抗体がマウスのイソ型であれば、マウス血清を免疫グロブリン/抗体のFc受容体への非特異的結合遮蔽に使用できる。精製PBMCには、10%ヒト血清が利用できる。 |
| 蛍光マイナス1(FMO) コントロール |
多色パネルで使用する全ての蛍光色素に対して、全蛍光色素よりそれぞれ1種類のみ減らして実験細胞を染色する。 |
検出チャネル内の蛍光分散の影響を検出するため(特により明るい蛍光色素)。 |
多色フローサイトメトリーパネルの構築に重要であり、ゲートをどこに設定するか決定する際に役立つ。 |
| 二次抗体コントロール |
二次抗体のみと培養した細胞。 |
二次抗体の非特異的結合を同定する。 |
二次抗体を使用する場合にのみ必要。 |
蛍光色素の最適化
蛍光分子は蛍光色素、フルオロフォア、蛍光色素としても知られ、多くの市販品をフローサイトメトリーに利用することができます。一次抗体にあらかじめ標識することでフローサイトメトリー解析がより簡便になります。以下のガイドをご参照ください。
◆ 多色パネルデザイン
- 最も明るい蛍光色素と低発現の抗原とを組み合わせる。抗原の発現レベルが不明な場合、より明るめの蛍光色素を用いることが望ましい。
- 蛍光色素間の流出やスペクトル重複を防ぐため、蛍光色素のスペクトラムを可能な限り分散させる。
- 1つ以上のレーザーで励起する蛍光色素(APC/Cy7など)は避ける。
- もし、3と4ポイント以上が得られない場合は、適切な補正コントロールを使用する。
- 染まり方が異なるサブ集団をより正確にゲートするため、FMOコントロールを使用する方がよい。
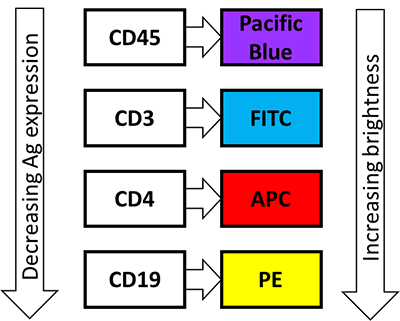
- 市販のオンライン多色パネルデザイナーの使用を考慮する。ポイント1〜3を描画する際、適切な4色パネルを使用する。下図のボックスの色は各蛍光色素を励起するレーザーに相当する。
細胞刺激最適化(細胞内サイトカイン染色用)
フロー実験を最適化するためには、細胞内サイトカインの産生を最大化するため、さらにin vitroで細胞刺激が必要な場合があります。このような目的によく使用される試薬としては、PMA, Ca++、ペプチド抗原決定基、タンパク質輸送阻害剤、ブレフェルジンAなどがあります。
下表に、標的タンパク質に対する刺激物とその培養時間を簡単に示しました。
- 分泌サイトカイン染色の場合、製造元の推奨濃度でブレフェルジンAまたはモネンシンを培養中に添加する。
- 未刺激物を少しよけておき、未染色コントロールと染色基底コントロールとする。
In vitro細胞刺激参照表
| 標的サイトカイン/リン酸化タンパク質 |
標的細胞 |
刺激物 |
処理時間 |
表面マーカー |
| IL-2 |
PBMCs |
PMA (50ng/ml) |
4-6 hours |
CD3 |
| IL-3 |
T-cells |
PMA(50ng/ml) + ionomycin (1µg(ml) |
4-6 hours |
CD4 |
| IL-4 |
PBMCs |
PMA(50ng/ml) + ionomycin (1µg(ml) |
4-6 hours |
CD4 |
| IL-6 |
PBMCs |
LPS (100ng/ml) |
4-6 hours |
CD14 |
| IL-10 |
PBMCs |
LPS (100ng/ml) |
18-24 hours |
CD14 |
| GM-CSF / IFNγ / TNFα / TNFβ |
PBMCs |
PMA(50ng/ml) + ionomycin (1µg(ml) |
4-6 hours |
CD3 |
| pStat5 |
PBMCs |
GM-CSF (20ng/ml) + IL3 (20ng/ml) |
15 min. |
CD123, CD116 |
| pStat3 |
PBMCs |
G-CSF (20ng/ml) + IL6 (20ng/ml) |
15 min. |
CD126, CD114 |
| pERK |
PBMCs |
IL3 (20ng/ml) + IL6 (20ng/ml) + FLT3L (20ng/ml) |
15 min. |
CD123, CD126, CD135 |
フローサイトメトリー解析における免疫染色の最適化
抗体を使用した染色手順には、抗体滴定、洗浄、ブロッキングなどの多くのステップが含まれます。最小限のバックグラウンドで最良の染色が得られる至適抗体濃度は実験的に決定する必要があります。抗体染色プロトコールの各ステップの最適化に以下のガイドをご参照ください。
- 製造元の指示に従って抗体の保存と取扱いを行い、崩壊とFc受容体介在性凝集を避ける。
- 高速遠心を4°Cで5分間行い、抗体凝集物を除去する。PE標識したIgM抗体は大きいため、本ステップは推奨しない。
- 抗体結合反応を氷上で直接光を避けて行う。
- 接着細胞を用いる場合、トリプシンが細胞表面抗原を切断する場合があるため、トリプシン処理は行わない。
- 抗体を希釈して分注保存する場合、染色緩衝液を希釈に用いる。0.09%アジ化ナトリウムを添加して細菌汚染を防ぐ。
- 各培養段階後に細胞を染色緩衝液で1回か2回洗浄し、結合していない抗体を除去する。
- 抗原が弱い場合にはシグナルを増大させるため、3ステップ染色過程を行うことも考慮する。この場合、ビオチン標識した一次抗体との抗原結合→ストレプトアビジン抱合型二次抗体との結合→蛍光色素に抱合した抗ストレプトアビジン抗体との最終結合、を行う。
- 死細胞や細胞堆積物を排除するため、抗体カクテルに生存率解析用試薬を添加する。
- 非特異的Fc受容体染色を防ぐため、Fc遮断ステップを行うか、FBSを染色緩衝液に添加する。または、イソ型コントロールを用いてFc受容体染色により得られたシグナルを差し引く。
- 蛍光色素のブリーチングを最小限に抑えるため、染色後、直ちに細胞を確認する方がよい。細胞を保存しなくてはならない場合、適切な固定液で固定し、4°Cで保存する。一晩保存する場合、0.5% PFA Iが好ましいが、複数日や複数週にわたるなどの長期では、エタノール固定を推奨する。
- 固定液に長期保存すると自己蛍光が顕著に増大するため、推奨しない。
- 最適なシグナルを得るために、抗体の正確な濃度を決定できるよう抗体滴定を推奨する。抗体の希釈率を種々試し、陽性コントロールで最も強いシグナルを与えネガティブコントロールで最も弱いシグナルを与える最低濃度において零点規正を行う。
お手元の抗体の濃度が不明な場合には、以下の推奨希釈率をご検討ください。
| 組織培養上清 |
腹水 |
全抗血清 |
精製抗体 |
| 1/100 |
1/1000 |
1/500 |
1 µg/mL |
フローサイトメトリー データ解析 - ゲーティング手順
フローサイトメトリーのデータ解析では、ゲーティングを基にして全体的な解釈をします。ゲートとは、解析対象の集団を定量し研究する上で、散乱またはマーカー発現のような共通した特徴をもつ細胞集団の周囲に配置した境界のことです。
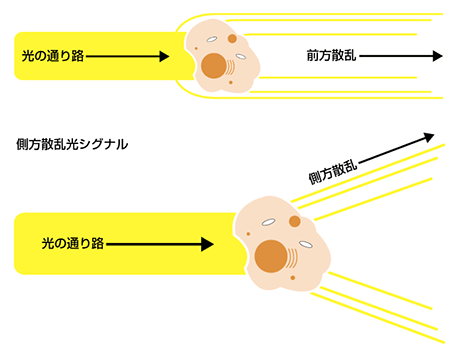
前方および側方散乱ゲーティング
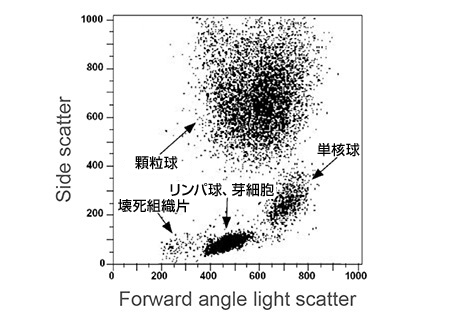
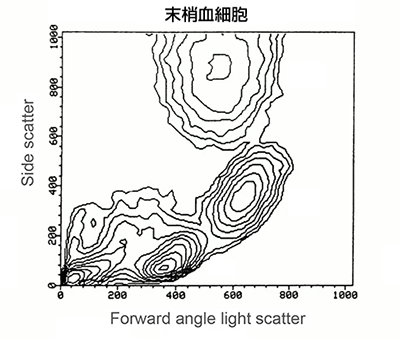
まず、細胞の散乱特性を基にしてゲートを行います。前方(FSC)および側方散乱(SSC)より、おおよその細胞の大きさと粒度がわかります。血液細胞集団の解析を例にとると、血液は様々な大きさや粒度の細胞から構成されていますので、散乱を基にしたゲーティングが非常に有用です(以下のドットプロットを参照)。赤色四角内の顆粒球はSSCが高いため、比較的容易に単球やリンパ球との境界を画定できます。黄色四角内の単球は、リンパ球(桃色四角)とは明らかに異なる集団を形成しており、より小さく粒度も低いです。紫色四角には壊死組織片とRBCsが含まれ、FSCとSSCの何れも最も低い値です。散乱光のゲーティングは、純度が問題にならず細胞を大量に選別する際に有用なツールです。
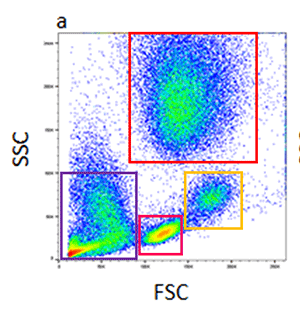
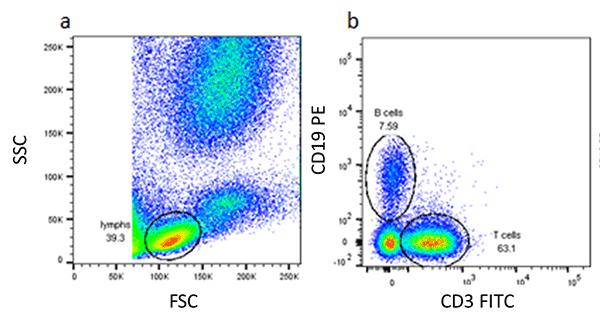
標識集団ゲーティング
散乱特性を基にして特定集団の境界を選択した次のステップでは、表面(または細胞間)マーカーを基準にしてさらに亜集団へと分割していきます。以下に示した典型的なドットプロットでは、FSCやSSCを基にしてリンパ球を最初にゲートし、その後、それぞれCD3やCD19の表面発現を基にしてT細胞とB細胞を分離します。右のドットプロットでは、T細胞とB細胞集団の明瞭な分界が見られます。
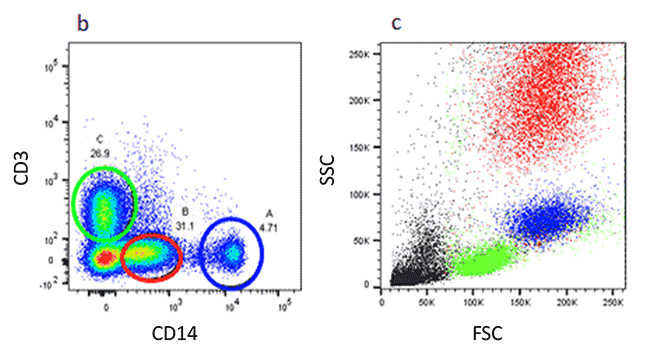
バックゲーティング
バックゲーティングは、ゲーティングパターンを確認する手法です。特定のゲートにより同定された集団を、完全に異なるパラメータで再度ゲートします。一般に、新しいゲーティング方法を試行している場合や非特異的染色や偽陽性の疑いがある場合に行われます
以下のドットプロットでは、血液細胞をまずCD3とCD14発現でゲートしてT細胞と非リンパ球とを分離しました。緑色、赤色、および青色で示した楕円は、それぞれT細胞、顆粒球、および単球を示しています。使用したゲーティング方法によるこれら集団の独自性を検証するため、FSCやSSCパラメータを用いて戻しゲーティングを行いました。右のドットプロットより、これらの散乱特性に基づいて各集団が確認できました。
*注:使用するグラフの種類や統計は解析ソフトウェアプログラムによって異なります。お使いのフローサイトメトリー機器のマニュアルに準じてどれを利用すべきかご検討ください。
フローサイトメトリー トラブルシューティングガイド
フローサイトメトリー(FACS)実験で、く遭遇する問題に関して、以下のガイドを原因の可能性と解決法のチェックリストとしてご利用ください。
蛍光強度が弱い、または蛍光シグナルが得られない。
| |
原因 |
解決策 |
| 1 |
抗体が分解または期限切れである。 |
- 抗体が製造元の使用説明書に準じて保存されていたことを確認する。
- 抗体ストックを記録し、期限切れでないことを確認する。
- 抗体の滴定を行い、同じ分注品を保存する場合は、保存バッファーに0.09%でアジ化ナトリウムを添加する。
|
| 2 |
蛍光色素の蛍光が衰えている。 |
|
| 3 |
抗体濃度が検出限度以下である。 |
- 使用前に抗体を滴定し、特定の実験に最適な使用量を決定する。
- 陰性(未染色)と陽性コントロールを使用する。
|
| 4 |
標的抗原の発現が低すぎる。 |
- 異なる細胞種における抗原発現を文献確認し、適切な陽性コントロールを使用する。
- 可能であれば、凍結サンプルではなく、新しく回収した細胞を使用する。
- 抗原(サイトカインなど)発現が事前のin vitro処理に依存する場合、細胞培養/刺激プロトコールを最適化する。
|
| 5 |
抗原-抗体結合が最適限界である。 |
- 抗体の生物種特異性を確認する。
- 抗体培養時間と温度を最適化する。
- ビオチン標識した一次抗体と追加のビオチン-ストレプトアビジンステップを利用し、シグナルを増幅することを考慮する。
|
| 6 |
細胞内抗原に接近できない。 |
|
| 7 |
細胞内抗原が分泌される。 |
- 標的を検出するためには、標的は膜結合型か細胞質に存在する必要がある。ブレフェルジンAなどのゴルジ遮断剤を利用する。
|
| 8 |
表面抗原が内部移行し始めている。 |
- プロトコールの全ステップを4°Cで行い、氷冷試薬を使用する。透過処理を氷上で行うことを推奨する。
|
| 9 |
染色細胞の蛍光がブリーチしている。 |
- 染色直後に細胞を確認する。
- 長期にわたり保存する場合は、固定液(PFAなど)をサンプルに添加する。アルコール固定液は避けた方がよい。
|
| 10 |
低発現抗原がほの暗い蛍光色素と対を形成している。 |
- 弱い抗原と、PEやAPCといった明るい蛍光色素を組み合わせる。
|
| 11 |
一次抗体と二次抗体との互換性がない。 |
- 一次抗体を産生した生物種で産生された二次抗体を使用する。
|
| 12 |
レーザーとPMTセッティングが蛍光色素と互換性がないか、蛍光特異的チャネルに対してPMT電位が低すぎる。 |
- データ取得前に適切な機器設定を読み込む。
- 全ての蛍光色素に対して設定を最適化するため適切な陽性・陰性コントロールを使用する。
|
| 13 |
蛍光シグナルが補正を超えている。 |
- 補正のために視覚的比較するのではなく、MFIアライメントを使用する。
|
飽和または過剰な蛍光シグナル
| |
原因 |
解決策 |
| 1 |
抗体濃度が高すぎる。 |
- 使用前に抗体を滴定し、特定実験に最適な使用量を決める。適切な陽性・陰性コントロールを使用する。
|
| 2 |
細胞内染色の場合、未結合の抗体が細胞に捕捉されている。 |
- 各抗体培養ステップ後に細胞を十分に洗浄し、TweenやTriton Xを洗浄バッファーに添加する。
|
| 3 |
高発現抗体と明るい蛍光色素が組合せられている。 |
- 強い抗原には、FITCやPacific Blueといったやや暗めの蛍光色素を組み合わせて使用する。
|
| 4 |
蛍光特異的チャネルに対してPMT電位が高すぎる。 |
- データ取得前に適切な機器設定であることを確認する。
- 全蛍光色素に対して設定を最適化するため、適切な陽性・陰性コントロールを使用する。
|
| 5 |
蛍光シグナルが補正以下である。 |
- 補正する際に視覚的比較ではなくMFIアライメントを使用する。
|
| 6 |
遮蔽が不十分 |
- ブロッキング段階段階だけでなく抗体とともに1%から3%の遮断剤を添加(遮断溶液に抗体を希釈)し、ブロッキング時間を延長する。
|
バックグラウンドが高いまたは非特異的染色
| |
原因 |
解決策 |
| 1 |
未結合の余剰な抗体がサンプルに存在する。 |
- 全ての抗体培養ステップ後に、細胞を十分に洗浄する。
|
| 2 |
非特異的細胞が標的されている。 |
- イソ型コントロールを使用し、Fc結合シグナルを差し引く。
- Fc遮断剤、BSA、またはFBSを用いて抗体培養前に細胞上のFc受容体を遮断する。
- 追加の洗浄を行う。
- 二次抗体抱合コントロールを使用して抱合体由来の非特異的シグナルを差し引く。
- 交差反応性のない二次抗体を選択する。
|
| 3 |
自家蛍光が高い。 |
- 未染色コントロールを使用し、自家蛍光シグナルを差し引く。
- 天然で自家蛍光の高い細胞(好中球など)には、自家蛍光が最小限(APCなど)である赤色チャネルで発光する蛍光色素を使用する。
- 上記解決策が不可能な場合、これらの細胞には非常に明るい蛍光色素を使用し、自家蛍光レベル以上のシグナルを増幅する。
- 固定液内での長期保存を避け、可能であれば染色後すぐに解析する。
|
| 4 |
死細胞が存在。 |
- データ取得前に細胞を一度ふるいにかけ、死細胞の堆積物を選別し除去する。
- PIや7-AADといった生存率色素を加え、死細胞を除外する。
- 可能であれば、凍結細胞ではなく新しく単離した細胞を使用する。
|
バックグラウンド散乱が高い、または異常な細胞散乱プロファイル
| |
原因 |
解決策 |
| 1 |
細胞の可溶化または損傷 |
- サンプル調製を最適化し、細胞の可溶化を防ぐ。
- 可能であれば、高速度での細胞のボルテックスや遠心を避ける。
- フレッシュなバッファーを使用する。
- 染色細胞を長期間保存することは避ける。
|
| 2 |
細菌感染。 |
- 染色細胞と抗体を適切に保存し、細菌増殖を避ける。細菌は低レベルの自家己蛍光を発する。
- 適切な無菌細胞培養を行い、細菌感染を防ぐ。
|
| 3 |
散乱には不適切な機器設定。 |
- 適切な機器設定であることを確認し、データ取得前に設定する。
- フレッシュで健康な細胞を使用し、細胞種に適したFSCやSSC設定を行う。
|
| 4 |
死細胞の存在。 |
- データ取得前に一度、細胞をふるいにかけ、死細胞堆積物を選別し除去する。
- 可能であれば、凍結細胞ではなく、新しく単離した細胞を使用する。
|
| 5 |
可溶化されていないRBCs(赤血球細胞)の存在。 |
- RBC細胞可溶化が完了したことを、顕微鏡下で確認する。
- 新鮮なRBC可溶化緩衝液を使用する。
- RBC堆積物を除去するため、必要な回数だけ洗浄する。
- フィコール密度勾配後にPBMCsを使用する場合、リンパ球中間相へのRBC汚染を最小限に抑えるための手順を最適化する。
|
異常なイベント・レート
| |
原因 |
解決策 |
| 1 |
細胞数が低いため、イベント・レートが低い。 |
- 最低細胞数を1X106/mlに維持する。
- 細胞を穏やかにピペッティングしてよく混合する。
|
| 2 |
サンプル凝集によりサンプルのイベント・レートが低い。 |
- データ取得前に細胞をふるいにかけ、堆積物を選別し除去する。
- 染色前に細胞を穏やかにピペッティングしてよく混合し、サンプル実行前に再度よく混合する。
|
| 3 |
機器設定が正しくない。 |
|
| 4 |
サンプルが注入チューブに詰まったため、事象が得られない。 |
- 製造元説明書に従ってフローサイトメトリー注入チューブの詰まりをとる(通常、10%の漂白剤を5-10分間流した後、dH2Oを5〜10分間流す)。
|
| 5 |
|
細胞培養を 1X106/mlに希釈する。 |
| 6 |
フローセル内またはシースフィルターの空気のため、事象率が非常に高い。 |
|
抗原決定基の損失
| |
原因 |
解決策 |
| 1 |
過剰なパラフォルムアルデヒド |
- パラフォルムアルデヒドが崩壊する際に放出するメタノールが染色に影響を及ぼす。1%のみのパラフォルムアルデヒドを使用する。
|
| 2 |
サンプルを氷上保管しなかった。 |
- 抗体は4°Cに保管して活性の損失を防ぐ。また、4°C保存することで、活性化脱リン酸化酵素やタンパク質分解酵素による対象抗原決定基の変更も防ぐことができる。
|
| 3 |
サンプルを長時間固定した。 |
- 固定プロトコールを最適化する。サンプルを長時間にわたり固定すると細胞損傷を引き起こすことがある。ほとんどの細胞は15分未満で固定できる。
|






















 このページを印刷する
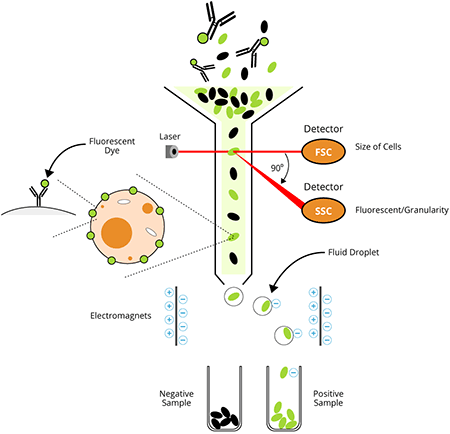
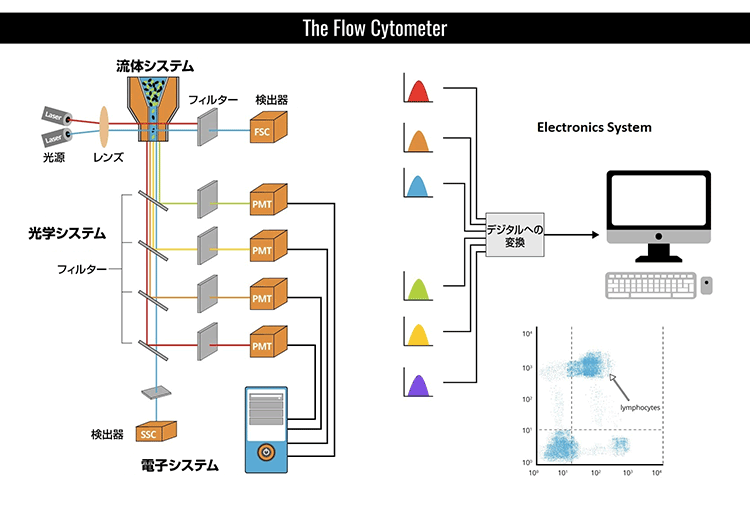
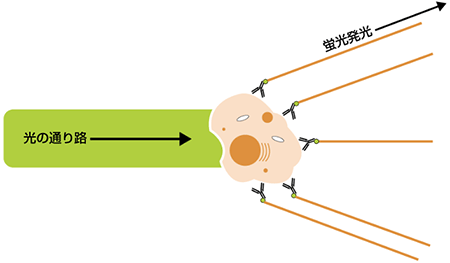
このページを印刷する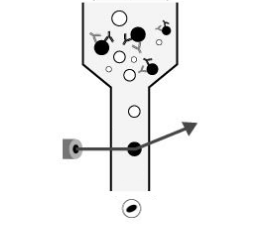 フローサイトメトリーは、多くのアプリケーションがある科学研究分野に不可欠な技術ですが、操作の基本原理はとてもシンプルです。細胞(あるいは他の粒子)を1列に並べてレーザーの前を通過させることで検出、計数、および選別を行います。このために、検出する粒子や細胞は、検出するフローサイトメトリーのレーザー波長に合った蛍光色素で標識されている必要があります。検出器のレーザーは、特定波長の光を放射する蛍光タグを励起します。フローサイトメトリーは、個々の細胞や粒子の物理的および科学的特性を迅速かつ同時にそして、複数のパラメーター分析ができる技術です。
フローサイトメトリーは、多くのアプリケーションがある科学研究分野に不可欠な技術ですが、操作の基本原理はとてもシンプルです。細胞(あるいは他の粒子)を1列に並べてレーザーの前を通過させることで検出、計数、および選別を行います。このために、検出する粒子や細胞は、検出するフローサイトメトリーのレーザー波長に合った蛍光色素で標識されている必要があります。検出器のレーザーは、特定波長の光を放射する蛍光タグを励起します。フローサイトメトリーは、個々の細胞や粒子の物理的および科学的特性を迅速かつ同時にそして、複数のパラメーター分析ができる技術です。