






















商品詳細
- 【1-01】 QuantSeq FWDとQuantSeq REVの違いは何ですか?
- 【1-02】 QuantSeqは分解されているRNAやホルマリン固定パラフィン包埋(formalin-fixed paraffin-embedded:FFPE)試料からもライブラリを調製できますか?
- 【1-03】 推奨のポジティブコントロールは何ですか?
- 【1-04】 QuantSeqのライブラリ調製において、もっとも重要な工程は何ですか?
- 【1-05】 QuantSeqのライブラリ調製にはどれくらいの時間がかかりますか?
- 【1-06】 QuantSeqライブラリ増幅のための推奨のPCRサイクル数はいくつですか?
- 【1-07】 QuantSeqでは、どの程度のマルチプレックス化が可能ですか?QuantSeqで使用されているバーコード(インデックス)システムについて教えてください。
- 【1-08】 QuantSeqと互換性のあるDual Indexing Kitはどれですか?
- 【1-09】 Lexogen UDI 12 nt Unique Dual Indexing Add-on KitによるDual PCRとQuantSeq FWD/REVで提供されているPCR Mixの違いについて教えてください。
- 【1-10】 ライブラリのサイクル数が足らなかった(アンダーサイクルとなった)場合の対処法を教えてください。
- 【1-11】 ライブラリのサイクル数が多すぎた(オーバーサイクルとなった)場合の対処法を教えてください。
- 【1-12】 バイオアナライザートレースでupper makerより大きなサイズの位置にあるピークは何を意味していますか?
- 【1-13】 QuantSeqライブラリの一般的なフラグメントサイズはどれくらいですか?
- 【1-14】 得られたQuantSeqデータは、データ解析パイプラインBlueBee®で解析できますか?
- 【1-15】 QuantSeqに含まれているデータ解析パイプラインBlueBee®のコードはどこにありますか?
- 【1-16】 internal primingを防ぐにはどうしたらいいでしょうか。
- 【1-17】 QuantSeqのデータ解析にマイコプラズマはどのような影響を及ぼしますか?
- 【2-01】 QuantSeq FWD HT kit(品番:015.384)は、通常のQuantSeq FWD kitと何が違うのでしょうか?
- 【2-02】 QuantSeq FWDライブラリに最適なシークエンスのリード長はどれくらいですか?
- 【2-03】 QuantSeq FWDに必要なRNAのインプット量はどれくらいですか?
- 【2-04】 QuantSeq FWDの最低インプット量はどれくらいですか?また、インプット量が少ない場合、プロトコールに変更点はありますか?
- 【2-05】 インサートサイズを長くする方法はありますか?
- 【2-06】 QuantSeq FWDライブラリに適したシークエンスプラットフォームは何ですか?
- 【2-07】 QuantSeq FWDライブラリでペアエンドシークエンスは可能ですか?
- 【2-08】 QuantSeq FWDのリードのシークエンスの読み取り方向は?
- 【2-09】 トリミングのステップでは何をトリミングするべきですか?
- 【2-10】 QuantSeq FWDのデータ解析に推奨されるアラインメントツールは何ですか?
- 【3-01】 QuantSeq FWDとQuantSeq REVの違いは何ですか?QuantSeq REVが必要なのはどのような場合ですか?
- 【3-02】 QuantSeq REVに必要なRNAのインプット量はどれくらいですか?
- 【3-03】 QuantSeq REVでのライブラリ調製にUMI Second Strand Synthesis Module(品番:081.96)を一緒に使用することはできますか?
- 【3-04】 QuantSeq REVライブラリに適したシークエンスプラットフォームは何ですか?
- 【3-05】 カスタムシークエンスプライマー(CSP)の使い方について教えてください。
- 【3-06】 QuantSeq REVに適したシークエンスのリード長はどれくらいですか?
- 【3-07】 QuantSeq REVライブラリでペアエンドシーケンシングはできますか?
- 【3-08】 QuantSeq REVのデータにpoly(T)伸長部位があり、シークエンスの品質が低いのですが、この理由を教えてください。
- 【3-09】 トリミングのステップでは何をトリミングするべきですか?
- 【3-10】 QuantSeq REVのデータ解析に推奨されるアラインメントツールは何ですか?
- 【3-11】 QuantSeq REVのシークエンスの読み取り方向は?
- 【4-01】 QuantSeq with UDI V1とV2では何が変わりましたか?
- 【4-02】 このキットで利用可能な最小サイズはどれくらいですか?
- 【4-03】 V1と V2を使用したQuantSeqで取得されたデータは同等ですか?
- 【4-04】 QuantSeq with UDI12(V2)は FFPEサンプルの分析に適していますか?
- 【4-05】 UDIシーケンスは、QuantSeq with UDI V1とV2で異なりますか?
- 【4-06】 QuantSeq with UDI12(V2)ライブラリーを増幅するには何回のPCRサイクルが必要ですか?
- 【4-07】 qPCRアッセイは、QuantSeq with UDI V1とV2で変わりましたか?
- 【4-08】 QuantSeq with UDI12(V2)ライブラリーからはどのようなサイズ分布が期待できますか?
- 【4-09】 QuantSeq with UDI12(V2)ライブラリーのサイクル数が不足している場合はどうすればよいですか?
- 【4-10】 QuantSeq with UDI12(V2)で取得したデータをLexogen data analysis solutionsで解析するにはどうすれば良いですか?
- 【5-01】バージョン 1 (V1) とバージョン 2 (V2) の違いは何ですか?
- 【5-02】V1 と V2 でキットの性能は変わりましたか?
- 【5-03】CORALL RNA-Seq V2 ライブラリの調製キットの付属品について
- 【5-04】CORALL V2 はどの種と互換性がありますか?
- 【5-05】RTMとRTLのどちらのプロトコルに従うべきですか?違いは何ですか?またどのように選べばよいですか?
- 【5-06】CORALL RTMとRTLで得られる結果は同等ですか?
- 【5-07】FFPE/低品質サンプルで RTL を使用できますか?
- 【5-08】なぜCORALL V2ライブラリのqPCRアッセイが必要なのですか?qPCRアッセイはどのように行うのですか?
- 【5-09】CORALL V2 ライブラリをシーケンスするにはどうすればよいですか?どのシーケンス形式を使用する必要がありますか?
- 【5-10】CORALL V2ライブラリをシーケンスに使用する時のローディング量はどのくらいですか?
- 【5-11】CORALL V2 シーケンスデータをどのように分析すれば良いですか?
- 【5-12】共有できる CORALL V2 データ分析パイプラインはありますか?
- 【5-13】CORALL mRNA-Seq V2 は、分解された RNA サンプルまたは FFPE RNA サンプルでの使用に適していますか?
- 【5-14】CORALL mRNA-Seq V2 ライブラリの調製に推奨されるinputは何ですか?
- 【5-15】CORALL mRNA-Seq V2は、分解されたRNAサンプルまたはFFPE RNA サンプルでの使用に適していますか?
- 【5-16】CORALL Total RNA-Seq V2ライブラリの調製に推奨されるinputは何ですか?
1. Lexogen社 QuantSeq 3' mRNA-Seq Library Prep Kit全般について
2.Lexogen社 QuantSeq 3' mRNA-Seq Library Prep (FWD)について
3.Lexogen社 QuantSeq 3' mRNA-Seq Library Prep Kit (REV)について
4.Lexogen社 QuantSeq 3' mRNA-Seq V2 Library Prep Kit FWD with Unique Dual Indices (12nt)[略称: QuantSeq with UDI12(V2)]について
5. Lexogen社CORALL RNA-Seq V2 Library Prep Kitについて
1. Lexogen社 QuantSeq 3' mRNA-Seq Library Prep Kit全般について
どちらのキットでも、mRNAの3'末端付近のcDNAが得られます。これらの違いは、Read1のリンカー配列の位置です。
FWDキットではRead1リンカー配列は第二鎖合成に使用するプライマーに組み込まれているため、シークエンスリードはpoly (A)尾部に向かって伸長します。QuantSeq FWDキットは遺伝子発現解析にお勧めです。
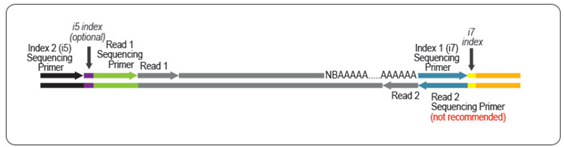
図1 QuantSeq FWDのシークエンス時の読み取り方向
QuantSeq REVキットではRead 1リンカー配列が第一鎖合成に使用するoligo(dT)プライマーの5'末端に位置しているため、転写終結点付近の正確な配列解析が可能です。QuantSeq REVキットでは、3'末端を直接読み取るために、キットに含まれるカスタムシークエンスプライマー(CSP)を用いてRead 1のシークエンス解析を行う必要があります。
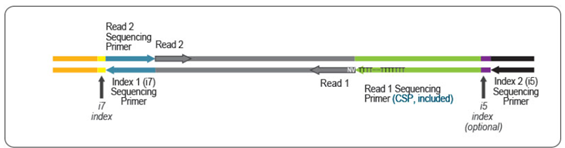
図2 QuantSeq REVのシークエンス時の読み取り方向
はい、QuantSeq FWDキットおよびQuantSeq REVキットの両方で、低品質RNAやFFPE試料を使用可能ですが、一部、下記に示すようなプロトコールの変更が必要になります。
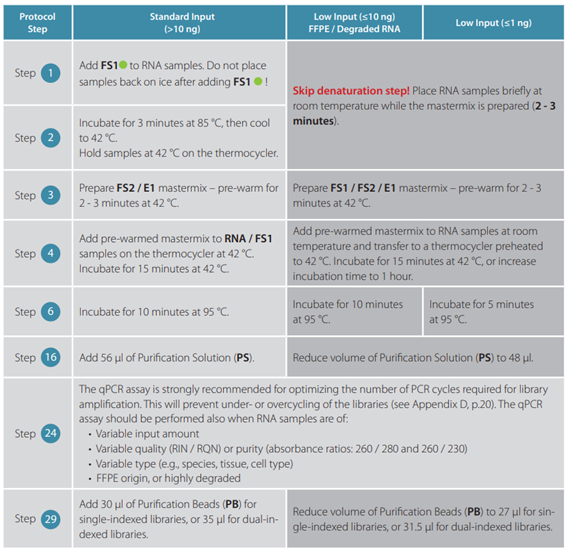
FFPE組織由来のRNA試料では、RNAの品質のばらつきが大きく、RIN値の精度が低いため、RIN値に加えてDV200値(200塩基以上のRNA断片の割合)を測定して、RNAの品質をご確認いただくことを推奨しています。また、FFPE組織由来のRNA試料では、ライブラリ増幅時のアンダーサイクルやオーバーサイクルを防止するために、qPCRアッセイを行って、PCRのサイクル数を最適化することを推奨しています。特に、遺伝子発現を比較したい場合、qPCRアッセイを行ってそれぞれのライブラリの平均サイクル数を決定し、最終的に比較するすべてのライブラリのサイクル数を同じにする必要があります。
すべてのQuantSeq製品でUniversal Human Reference RNA(アジレント・テクノロジー株式会社)をポジティブコントロールとして使用することを推奨しています。
QuantSeq FWD/REVのライブラリ調製で最も重要な工程は、第一鎖合成です。サンプルの温度を42℃に保つことで、oligo(dT)プライマーのpoly(A)尾部へのハイブリダイゼーションが確実になり、ミスハイブリダイゼーションを防ぐことができます。QuantSeqを初めてご使用される場合は、第一鎖合成のサポートビデオをご視聴いただくことを推奨しております。
4.5時間で total RNA から 3' mRNA-Seq ライブラリを調製可能です。QuantSeqを初めてご使用される場合、PCRのサイクル数を最適化していただく必要があり、qPCRを行う時間(約2.5時間)含め、余裕をもって実験を行っていただくことを推奨しております。また、実験前にUser Guideをご一読いただき、各試薬が正しく準備されていることを事前にご確認ください。
最適なサイクル数は、サンプルタイプ(由来、組織、RNA品質等)によるため、qPCRアッセイで決定していただくことを推奨しておりますが、目安としてUniversal Human Reference RNA (UHRR)のインプット量と最適サイクル数(下記の表)をご参考ください。

QuantSeqでは高度なマルチプレックスが可能です。インデックスはエンドポイントPCR中にバーコードとして導入されます。通常のQuantSeqには最大96個の6 nt i7インデックスが含まれています。Dual Indexをご希望の場合、4種類または96種類の6 nt i5インデックスが含まれる、別売りのLexogen i5 6 nt Unique Dual Indexing Add-on Kitを組み合わせることで、最大9216種類のバーコードを使用することができます。
Unique Dual Index (UDI) でインデックスホッピングの影響を抑えたい場合は、最大384種類のpre-mixed UDIsが含まれる、別売りのLexogen UDI 12 nt Unique Dual Indexing Add-on Kitを組み合わせることで、インデックスエラーを修正することができます。また、最大384種類の12 nt UDIがあらかじめ含まれているQuantSeq 3' mRNA-Seq Library Prep Kit (FWD) with UDI 12 ntのご用意もございます。
QuantSeq-FWD, QuantSeq-REV, QuantSeq-Flex, QuantSeq-Pool ライブラリ調製キットは下記のDual Indexing Kitと互換性があります。
| 品番 | 品名 | 包装 |
|---|---|---|
| UDI 12 nt Unique Dual Indexing Add-on Kits contain primer plates and PCR Enzyme and PCR Buffer | ||
| 107.96 | Lexogen UDI 12 nt Unique Dual Indexing Add-on Kit, Set A1 (UDI12A_0001-0096), 1 rxn/UDI | 96 PREP. |
| 108.96 | Lexogen UDI 12 nt Unique Dual Indexing Add-on Kit, Set A2 (UDI12A_0097-0192), 1 rxn/UDI | 96 PREP. |
| 109.96 | Lexogen UDI 12 nt Unique Dual Indexing Add-on Kit, Set A3 (UDI12A_0193-0288), 1 rxn/UDI | 96 PREP. |
| 110.96 | Lexogen UDI 12 nt Unique Dual Indexing Add-on Kit, Set A4 (UDI12A_0289-0384), 1 rxn/UDI | 96 PREP. |
| 111.96 | Lexogen UDI 12 nt Unique Dual Indexing Add-on Kit, Set B1 (UDI12B_0001-0096), 1 rxn/UDI | 96 PREP. |
| 120.96 | Lexogen UDI 12 nt Unique Dual Indexing Add-on Kit, Sets A1-A4 (UDI12A_0001-0384), 1 rxn/UDI | 384 PREP. |
| i5 6 nt Indexing Add-on Kits | ||
| 047.4x96 | Lexogen i5 6 nt Dual Indexing Add-on Kit (5001-5004), 96 rxn/Index | 96 PREP. |
| 47.96 | Lexogen i5 6 nt Unique Dual Indexing Add-on Kit (5001-5096), 1 rxn/Index | 96 PREP. |
【1-09】 Lexogen UDI 12 nt Unique Dual Indexing Add-on KitによるDual PCRとQuantSeq FWD/REVで提供されているPCR Mixの違いについて教えてください。
QuantSeq FWD/REVで提供されているPCR Mixには、インデックスの無いP5プライマーが含まれています。そのため、このPCR Mixをi5およびi7インデックスプライマーと一緒に使用すると、最終的に得られたライブラリにはi5インデックスを持たないインサート断片が一部存在することになります。このリードはdemultiplexing中に失われます。12 nt UDIを使用する場合は、QuantSeq 3' mRNA-Seq FWD with UDI 12 nt KitsのUser Guide(113UG227)に記載されている、Endpoint PCR プロトコールに従ってください。
大抵、アンダーサイクルとなってしまったライブラリでもプーリングとシークエンスに十分な量のcDNAが含まれているため、再度PCRを行う必要はありません。ライブラリの量がシークエンスに十分かどうかは、Lexogen社のライブラリ定量計算ファイルで確認できます。
このファイルに測定した濃度を入力し、プールする各ライブラリの量を調節します(最小量は10 fmol)。プールに必要な量がアンダーサイクルで得られたライブラリのcDNAの量を下回っていれば、ライブラリはシークエンス可能です。シークエンスに十分な量が得られなかった場合、別売りのPCR Add-on Kit for Illumina, 96 rxn(品番:020.96)に含まれる再増幅プライマーを使用してPCRを行う必要があります。PCR Add-on Kit for Illumina, 96 rxnは、single indexライブラリにのみ使用できる点ご注意ください。dual indexライブラリは別売りのReamplification Add-on Kit for Illumina(品番:080.96)でのみ再増幅が可能です。
バイオアナライザートレースにおけるベースラインの上昇(下記図参照)や、1000〜9000 bpにあるセカンドピークはオーバーサイクルの兆候です。
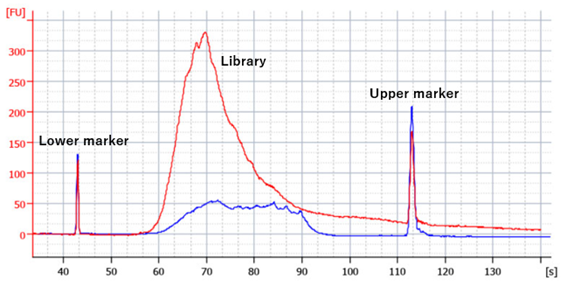
図3 適切なサイクル(10 ng UHRR, 19 cycles,青)とオーバーサイクル(10 ng, 24 cycles,赤)で作製したQuantSeqライブラリのバイオアナライザートレース
オーバーサイクルの原因は、ライブラリ増幅のPCRサイクル数が多すぎて、PCRでプライマーとテンプレートが枯渇し、生成された二本鎖cDNAが変性し始め、誤って再アニーリングされてしまった際に生じます。これにより、バイオアナライザーチップやゲル上を低速で流れる長くて大きな分子が生成され、バイオアナライザーだけではライブラリの正確な定量ができなくなってしまいます。
オーバーサイクルが疑われるライブラリを正確に定量するには、バイオアナライザーまたは同様の分析に加えて、qPCRアッセイを行う必要があります。発現データの偏りを無くすための最善の方法は、ライブラリを再調製し、ライブラリの増幅時のPCRサイクル数を減らす方法です。ライブラリの再調製が難しい場合(貴重なサンプルなど)は、オーバーサイクルによりPCR duplicates が多くなり、定量精度やクラスター分析に影響を与える可能性があるものの、シークエンスのステップに進むのが最善です。
オーバーサイクルを防ぐには、qPCRアッセイを行ってライブラリ生成に必要な最適なPCRサイクル数を決定する必要があります。同じようなサンプルであれば、ライブラリ増幅(エンドポイントPCR)のPCRサイクル数を減らすことでオーバーサイクルを防ぐことができます。オーバーサイクルライブラリの収量に応じて減らすべきサイクル数は下記を参考に決定してください。
| 半分の収量に減らしたい場合 | 1サイクル |
| 4分の1の収量に減らしたい場合 | 2サイクル |
| 10分の1の収量に減らしたい場合 | 3サイクル |
upper makerより大きいサイズの位置で見られるベースラインの上昇は、オーバーサイクルを示している可能性があります(詳細はQ【1-11】をご確認ください)。 または、精製ビーズの混入(ビーズのキャリーオーバー)が原因で、upper maker周辺やそれより大きな位置にピークが生じる可能性があります。ビーズのキャリーオーバーを防ぐために、溶出液を回収する際は、サンプルをマグネット上において、5分ほどインキュベートした後、ビーズに溶出液を2 µlほど残すようにしてください。サンプル全量を回収しないように、15〜17 µlのみを新しいPCRプレートまたはチューブに移してください。
QuantSeqのプロトコールは、短いリード(SR75〜100)用に最適化されており、平均ライブラリサイズはおよそ335〜456 bp、平均インサートサイズは203〜324 bpです。別売りのQuantSeq-Flex First Strand Synthesis Module V2 for Illumina(品番:166.96)をQuantSeqと一緒に用いることで、QuantSeqライブラリのリード長を延長することができます。詳細についてはUser Guideをご確認ください。
すべてのQuantSeq FWD/REVキットには、無料のデータ解析パイプラインBlueBee®にアクセスするためのコードが付属しています。提供されるパイプラインはキットのPREP数分だけ含まれています。例えば96 PREPのキットの場合、96 PREP分のデータ解析を実行できます。BlueBee®で解析可能な適応種は、こちらをご確認ください。
QuantSeq FWD/REVキットに含まれている無料のBlueBeeRコードは、最大1.5 GBまでのFASTQファイルに対応しています。それ以上の大きいサイズのデータを解析したい場合は、ファイルを圧縮するか、大きなサイズのファイルに対応したBlueBeeRコードを別途ご購入いただく必要がございます。これらの詳細についてはこちらをご参照ください。
QuantSeq FWDで提供されるコードには、下記の2種類のパイプラインオプションがございます。
- FWD pipeline:標準のQuantSeq FWDライブラリデータ用
- FWD-UMI pipeline:別売りのUMI Second Strand Synthesis Module for QuantSeq FWD for Illumina(品番:081.96)を使用して作成されたQuantSeq FWDライブラリ専用
すべてのQuantSeq FWD/REVキットにはBlueBee®のアクティベーションコードが付属しています。このコードはメインのQuantSeqキットのボックスに入っている小さな試薬ボックスの側面に貼られたステッカーに印刷されています。

QuantSeq FWD with UDI kitsでは、Library Generation Module試薬ボックス内のカードにアクティベーションコードが記載されています。
マイコプラズマRNAがQuantSeqライブラリ調製のインプットとして使用されるサンプルに存在する場合、全体的なマッピング率が低下して、標的遺伝子における特異的なマッピング率が低下してしまいます。最悪な場合は30%以下のマッピング率になることがあります。また、リードをマイコプラズマにマッピングすると、固有のマッピングリードを特定することができます。異なるマイコプラズマ種が存在している可能性があるため、BLASTnを使用してシークエンスを確認してください。

2. Lexogen社 QuantSeq 3' mRNA-Seq Library Prep (FWD)について
QuantSeq FWD HT kit(品番:015.384)は、96種類のi7インデックスを含む標準のQuantSeq FWD kitに、4種類のi5インデックスが追加され、最大384種類のライブラリ調製が可能な高い処理能力を備えたキットです(high-throughput version:HT)。index hoppingを押さえたい場合は、あらかじめ384種類のUnique Dual Index(UDI)を含む、QuantSeq 3' mRNA-Seq Library Prep Kit FWD with UDI 12 nt Sets A1-A4(品番:115.384)がお勧めです。
ほとんどのアプリケーションにおいて、single-read 50 bp (SR50) または single-read 75 bp (SR75) で十分です。ライブラリにUnique Molecular Indentifiers(UMI)が含まれている場合は、推奨の最小リード長はSR75 です。 また、ゲノム配列の1か所にのみマッピングされるリード(ユニークマッピングリード)を増したい場合や、poly(A)尾部まで読み取ることで3'末端の配列を正確に解析したい場合は、より長いリード長(SR100, SR150)が適用可能です。
サンプル中に含まれるpoly(A)RNAの含有量に依りますが、推奨のRNAインプット量は1〜500 ngです。(total RNAのインプット量が10 ng未満の場合は一部プロトコールの修正が必要です。(Q【2-04】 参照)total RNAのpoly(A)濃縮や、rRNA除去は必要なく、FFPE組織由来のRNAのような、低品質のRNAもご使用いただけます。低発現量の転写産物を効率的に検出するためには、200 ng以上のインプット量を推奨しており、最初はtotal RNAのインプット量を500 ngでお試しいただくことをお勧めいたします。
インプット量が1 ng未満でも機能しますが、10 ng以下のインプット量で実験する場合は、total RNAのインプット量に応じて、下記のプロトコールの変更が必要です。
QuantSeq FWDと併せて、別売りのQuantSeq-Flex First Strand Synthesis Module(品番:166.96)をご使用いただくことで、より長いインサートサイズを持つライブラリが調製可能です。手順の詳細については、当該商品のUser Guideをご確認ください。
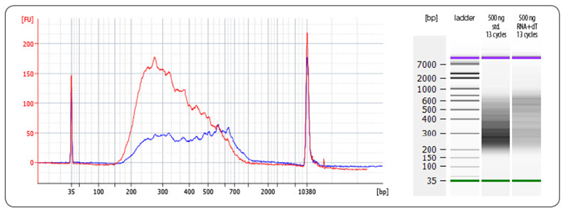
図4 QuantSeq-Flex First Strand Synthesis Moduleを用いて得られた、より長いインサートサイズのライブラリの例
標準のQuantSeq(赤色、RNA+dT)およびQuantSeq-Flex(青色、std.)を用いてUniversal Human Reference RNA(UHRR)500 ngから得られたサンプルのバイオアナライザートレースを示した。インプットRNAは標準のQuantSeqのFS1 bufferまたはQuantSeq-Flexの5 µl oligo(dT)を用いて85℃で3分間変性させた。RNA+dT条件下では、平均ライブラリサイズが増大する。ライブラリはi7およびi5のインデックスプライマーを用いて、Lexogen i5 6 nt Dual Indexing Add-on Kit(品番:047.4X96、047.96)に含まれるDual PCR Mixでサイクル数:13で増幅した。
QuantSeq FWDキットは、下記のIllumina社機器でのシークエンス解析に適しています。
- GAIIX
- iSeq 100
- HiSeq 2000/2500/3000/4000
- MiSeq
- NextSeq 500/550/2000
- MiniSeq
- NovaSeq
QuantSeq FWDライブラリにおけるRead 2は伸長開始時にpoly(T)配列があるため品質が非常に低く、ペアエンドシークエンスは推奨しません。もし、ペアエンドシークエンスモードでシークエンス処理をした場合は、Read 2のデータは破棄し、その後の解析にはRead 1のみを用いるようにしてください。ペアエンドシークエンスにはQuantSeq REVを推奨しております。
QuantSeq FWDのリードは1 転写産物あたり 1分子の断片のみ逆転写されます。そのため、QuantSeq FWDでは主に3'末端付近を解析し、Read 1の配列はmRNAの配列を直接反映しています。
QuantSeqのシークエンスリードでは、アダプター配列、poly(A) / poly(T)配列、および低品質ヌクレオチドを除去する必要があります。また、短すぎるリード(<20 nt)、または低品質なリードも、アラインメントの前に除去する必要があります。第二鎖合成ステップはランダムプライミング法に基づいているため、ランダムプライマーがcDNAへ非特異的にハイブリダイゼーションし、インサートの最初のヌクレオチドのエラーが起こりやすくなります。そのため、QuantSeq FWDのデータの場合、アラインメント中にリードエンドのsoft-clippingが実行できる、STAR alignerのようなアラインメントツールを用いて、許容ミスマッチ数を14まで増やすことを推奨しております。あるいは、HISAT 2のような、より厳密なアラインメントツールを使用する場合、アラインメントの前にRead 1の最初の12 ntをトリミングすることを推奨いたします。リードをトリミングすると、アラインメントに適した長さのリード数を減らすことができますが、リードの品質向上によりマッピングリードの絶対数が増加する可能性があります。
QuantSeq FWDデータのマッピングにはSTAR alignerを推奨しています。リードは最後のエクソン部位に到達せず、連結部分をまたぐこともあります。STAR alignerでは、アラインメント中にリード端のsoft-clippingを実行できるため、アラインメント率を最大限に高めることができ、アラインメント前に追加の5'ヌクレオチドをトリミングする必要がなくなります。ただし、poly(A)尾部とシークエンスアダプタを除去するには、アラインメントの前に必ずリードのトリミングが必要です(Q【09】参照) 。
3. Lexogen社 QuantSeq 3' mRNA-Seq Library Prep Kit (REV)について
どちらのキットでも、mRNAの3'末端付近のcDNAが得られます。これらの違いは、Read1のリンカー配列の位置です。QuantSeq REVではRead 1リンカー配列が、oligo(dT)プライマーの5'末端に位置し、Read 2リンカー配列が第2鎖合成中に使用されるランダムプライマーの5'末端に位置しているため、QuantSeq FWDとは逆方向のライブラリが生成されます。
FWDキットではRead1リンカー配列は第二鎖合成に使用するプライマーに組み込まれているため、シークエンスリードはpoly(A)尾部に向かって伸長します。FWDキットは遺伝子発現解析にお勧めです。
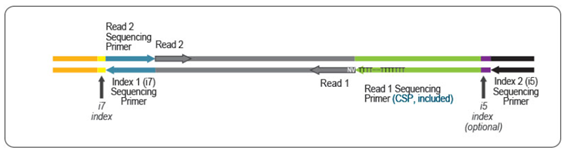
キットに含まれるカスタムシークエンスプライマー(CSP)は、Read 1の開始時にpoly(T)伸長シークエンスを避けるために、QuantSeq REVライブラリのシークエンスに必要で、これによりRead 1が正確に転写産物の3'末端から開始されます。
QuantSeq REVではペアエンドシークエンスで3'UTRアイソフォームを同定するのに有効で、選択的ポリアデニル化部位の同定や、3'UTRアイソフォーム発現研究などにお勧めです。
推奨のtotal RNAインプット量は、10〜500 ngです。QuantSeq FWDとは異なり、最小インプット量は10 ngで、低インプット量に対応したプロトコールは適用できません。また、total RNAのPoly(A)濃縮やrRNA除去は必要ありません。
QuantSeqに必要なtotal RNA量はサンプルにおけるpoly(A)RNA含有量によって異なります。Lexogen社では様々な培養細胞、動物や植物組織、酵母、真菌、ショウジョウバエ、およびヒトReference RNAにて検証済です。低量の転写産物を効率的に検出するためには、200 ng以上のインプットRNAが推奨されるため、最初は、500 ngのtotal RNAでお試しいただくことを推奨しています。インプット量に限りがある場合は、より長いシークエンスリード長をご選択いただくことで、3'末端の読み取りも可能ですので、QuantSeq FWDをお試しいただくことを推奨いたします。
UMI Second Strand Synthesis Module(品番:081.96)はQuantSeq FWD のみに対応しているため、QuantSeq REVではご使用いただけません。UMI Moduleに含まれる、UMIを持つランダムプライマーに、QuantSeq REVで使用されるoligo(dT)プライマーのP5アダプター配列が含まれるため、QuantSeq REVでUMI Moduleを使用しても、P7アダプターが無く、適切なライブラリができません。
QuantSeq REVでは、Read 1の配列決定にカスタムシークエンスプライマー(CSP)を必要とするため、CSPが適用可能なすべてのIlluminaプラットフォームでシークエンス可能です。これらのプラットフォームとして、NextSeq 500/550および、NextSeq 2000、HiSeq 2000/2500/3000/4000、NovaSeq 6000、MiSeq計測器が含まれます。QuantSeq REVライブラリはCSPに対応していないiSeq 100とは互換性がありません。
QuantSeq REVで提供されている、CSP Version 5 (CSPV5)は、poly(T)伸長部位をカバーし、Multiplex Read 1 Sequencing Primerとして機能します。 各Illumina機器におけるCSPV5の正しい使用方法については、こちらをご参考ください。なお、iSeq 100では、CSPV5は使用できません。
QuantSeq REVのシークエンスでは、single-read 75-150 bp(SR75〜150)で読み取ることが好ましく、読み取りは正確に3'UTRから始まるため、読み取りの最初は転写産物の末端から25 ntまでをカバーするポリアデニル化シグナル部位を含んでいます。 ポリアデニル化シグナルは全ての転写産物で非常に類似しているため、より長いリードほど、ユニークマッピングの比率が高くなります。
はい、QuantSeq REVではペアエンドシーケンシングが実行可能です。QuantSeq REVでは転写産物における3'末端の正確な読み取りの為に、Read 1用のカスタムシークエンスプライマー(CSP)が必要です。
考えられる理由として下記の2つが挙げられます。
- Read 1のシークエンスにカスタムシークエンスプライマーVersion 5(CSPV5)を使用していない。
CSPV5を使用しないと、Read 1の開始時にpoly(T)伸長が行われ、読み取りの精度と実行結果が損なわれます。ご使用のシーケンシング機器が、CSPに対応していることをご確認いただき、こちらから、正しい手順でCSPが使用されていることをご確認ください。 - ライブラリ調製中における、第一鎖合成中にoligo(dT)プライマーがミスハイブリダイゼーションした。
ライブラリ調製時に、サンプルを42℃に維持していないと、oligo(dT)プライマーがミスハイブリダイゼーションしてしまう恐れがあります。これにより、インサートの短いライブラリが転写産物のpoly(A)尾部にさらにプライミングされてしまい、CSPV5を使用しているにも関わらず、Read 1の開始時に長いpoly(T)伸長部位を持つリードが発生する可能性があります。ライブラリ調製時のミスハイブリダイゼーションを防ぐための、第一鎖合成の注意事項については第一鎖合成のサポートビデオを参照ください。
QuantSeq REVを用いて行ったシングルリードシークエンスではトリミングの必要はありませんが、ペアエンドシーケンシングでは、Read 2の最初の12ヌクレオチドのトリミングが必要です。別の方法としては、STAR alignerを使用して、許容ミスマッチ数を16に設定することを推奨しております。アダプターの混入を検出する場合、リードをアラインメントするためにcutadaptやtrim-gallore、bbdukなどでトリミングしていただくことを推奨しています。
QuantSeq REVデータのマッピングにはSTAR alignerを推奨しています。リードは最後のエクソン部位に到達せず、連結部分をまたぐこともあります。poly(A)尾部とシークエンスアダプタを削除するには、アラインメントの前に必ずリードのトリミングが必要です。QuantSeq REVのデータ分析の詳細については、こちらをご参考ください。
Read 1の読み取りは、poly(A)尾部から開始し、転写配列を再び読み取ってcDNA配列を反映します。そのため、Read 2はmRNAの配列を直接反映し、poly(A)尾部に向かって転写されます。
4. Lexogen社 QuantSeq 3' mRNA-Seq Library V2 Library Prep Kit FWD with Unique Dual Indices (12nt)[略称: QuantSeq with UDI12(V2)]について
QuantSeq 3' mRNA-Seq V2 Library Prep Kit FWD with Unique Dual Indices (12nt) (V2)はQuantSeq 3' mRNA-Seq Library Prep Kit (FWD) (V1)の後継品です。V2のV1との主な違いは新しく改良されたPCRシステムが導入されていることです。
V2キットでは、V1キットに含まれていたDual PCR Mix「Dual PCR」とEnzyme Mix「E」が、PCR Mix「PM」とPCR Enzyme Mix「PE」に置き換えられています。PCR構成品の変更に伴い、PCR プログラムも以下のようにアップデートされました。
・アップデート前のQuantSeqのPCRプログラム (V1)
・アップデート前のQuantSeqのPCRプログラム (V2)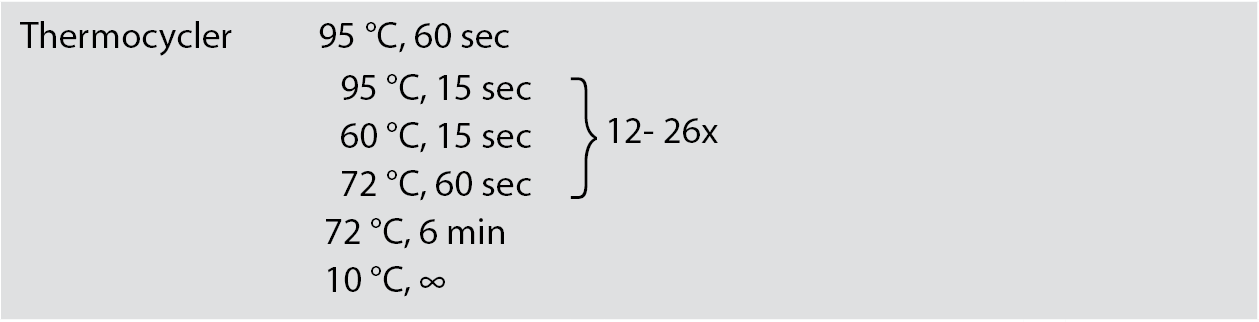
はい。 Unique Dual Indices (12nt) (V2)を備えた QuantSeq 3 ' mRNA-Seq V2 Library Prep Kit FWD と Unique Dual Indices(V1)を備えた QuantSeq 3' mRNA-Seq Library Prep Kit FWDで得られたデータの間には高い相関関係があることを確認しています。
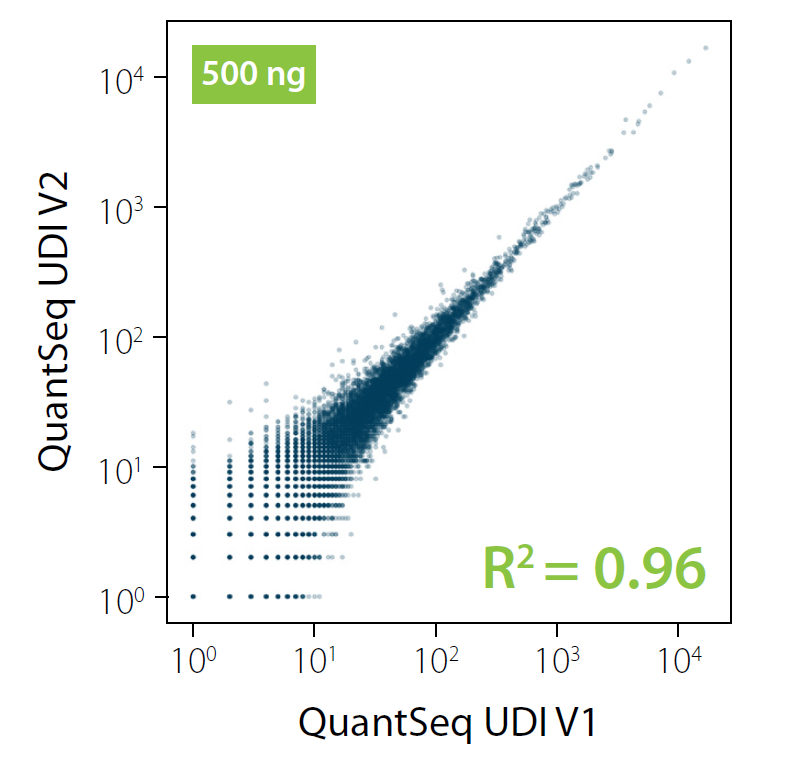
図5 500 ngのUHR + SIRV3コントロールセットで得られた相関プロット
QuantSeq UDI 「V1」(x軸)と「V2」(y軸)を比較した。
はい。低品質サンプルやFFPE サンプルも、UDI12 (V2)のQuantSeqで解析できます。QuantSeq with UDI12 V2で、新鮮凍結(FF)サンプルおよびホルマリン固定パラフィン包埋(FFPE)サンプルを解析したシーケンスデータを比較から、検出される遺伝子の高い重複を確認しています。
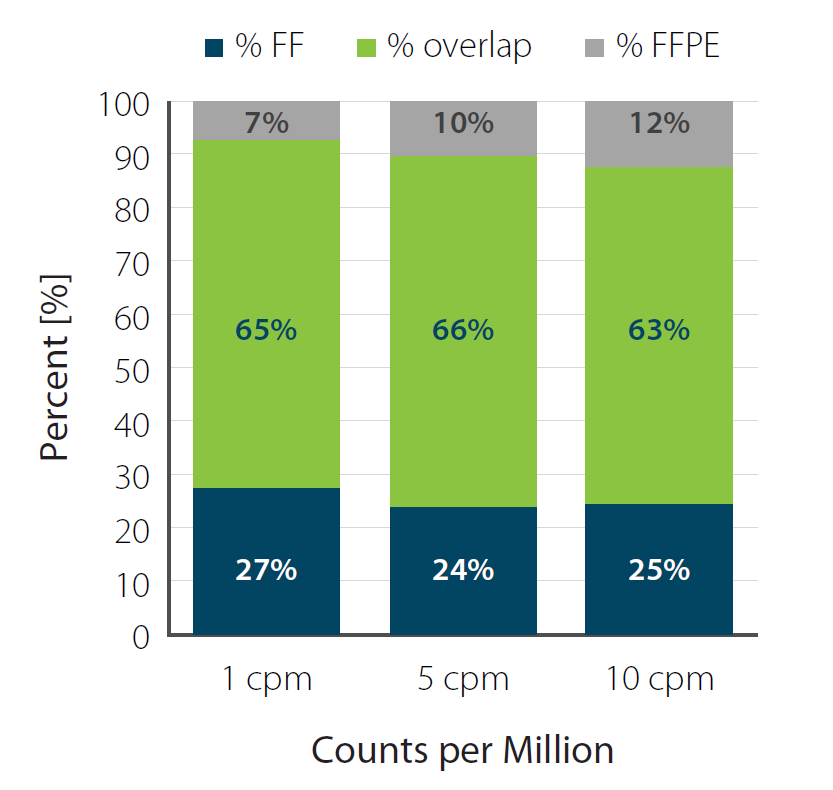
図6 凍結(FF)あるいはホルマリン固定パラフィン包埋(FFPE)したマウス脾臓サンプルから抽出したRNAを用いたシーケンスデータ
input RNA: 10ng, リード:1M
横軸は左から1cpm、5cpm、10cpm。
RIN値※1:3.7(FF)および2.9(FFPE)。DV200値※2:90%(FF)および 9% (FFPE)
※1 RIN値:RNA分解の指標。1〜10段階で、1に近いほどRNAの分解が進んでいることを表している。
※2 DV200値:測定したRNA中に200塩基以上のRNA断片が存在する割合。数値が低いほどRNAの断片化が進んでいることを表している。
いいえ。UDIセットのA1〜A4およびB1のシーケンスは、QuantSeq with UDI V1 とV2で使用されているものは同じです。これらの配列は、Lexogen UDI 12 nt Unique Dual Index Sequences ファイルから確認できます。
最適なPCR サイクル数は、サンプルの品質と由来によって異なります。そのため、qPCR アッセイで決定する必要があります。
qPCRアッセイには以下が必要です。
・PCR Add-on Kit for Illumina (品番:020.96)
・[SYBR Green I nucleic acid stain (10,000X in DMSO, DMSOはキットに不含)], [Sigma-Aldrichの品番:S9430]または[ThermoFisherの品番:S7585]を推奨します。
注意事項:他のSYBR Green IやqPCR mastermixesの使用は推奨しておりません。
プロトコル
qPCRアッセイに推奨されるプロトコルは、PCR Add-on Kit Instruction Manual とQuantSeq 3' mRNA-Seq Library Prep Kit ユーザーガイドの付録に記載されています。
Additional Resources
qPCRアッセイの詳細については、以下のFAQを参照してください。
・What do I need for the qPCR Assay?
・How do I calculate the Endpoint PCR cycle number from the qPCR assay?
・How many PCR cycles are needed to amplify QuantSeq libraries?
いいえ。qPCR アッセイ、qPCRプロトコル、およびendpoint PCRの正しいサイクル数の決定に必要な試薬は、UDI V1 および V2のQuantSeqで同じです。
QuantSeq with UDI12(V2) ライブラリーの典型的な分布は、inputサンプルの種類とinput量に依存します。例えば、FFPEサンプルは通常high-quality Universal Human Reference RNA (UHRR)よりも短いライブラリーを生成します。以下は、ユーザーガイド に従い(あるいは一部プロトコルの変更を加えて)、UHRR RNAをQuantSeq with UDI12(V2)で解析した時のライブラリー分布例です。
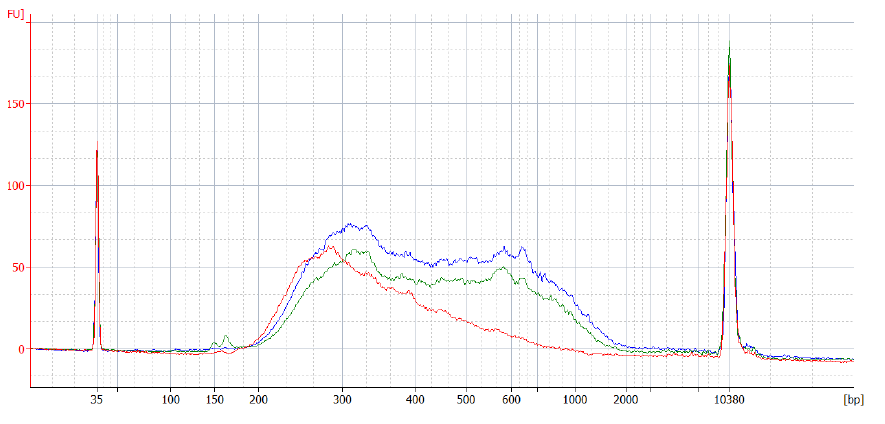
図7 異なるinput量のUHRRから調製されたQuantSeq FWDライブラリーに対する、Bioanalyzerによるサイズ分布のトレース
赤:UHRR RNAを500 ng使用。ライブラリーは標準プロトコルに従い作製。青, 緑:First Strand cDNA synthesisの初期ステップを省略するlow input用のプロトコル(詳細はこちら )に従いライブラリーを作製。青では10 ng、緑では1 ngのUHRR RNAを使用。Endpoint PCRは独自のdual indicesを使用して実行した。
Dual-indexed librariesは、Reamplification Add-on Kit for Illumina(品番:080.96)を使用することで再増幅できます。
2023 年 1 月以降、BlueBee® Genomics Platform上での QuantSeqのデータ分析は廃止されました。データ分析の継続性を確保するために、Lexogen社は独自のバイオインフォマティクスデータ分析プラットフォームを開発しており、間もなく利用可能になる予定です。このプラットフォームが開始されるまで、Lexogen社は社内パイプライン (BlueBee® data analysisのベースとする) を使用してお客様のデータを分析します。有効なアクティベーションコードをお持ちのお客様は、これを無料でご利用いただけます。
5. Lexogen社CORALL RNA-Seq V2 Library Prep Kitについて
CORALL V2キットにはRTMとRTL という2種類の逆転写試薬が付属しています。RTM試薬は平均約350bp(赤色のトレース)のライブラリを生成しますが、RTL試薬を使用すると平均ライブラリサイズを約550bp(青色のトレース)に増加させることができます。
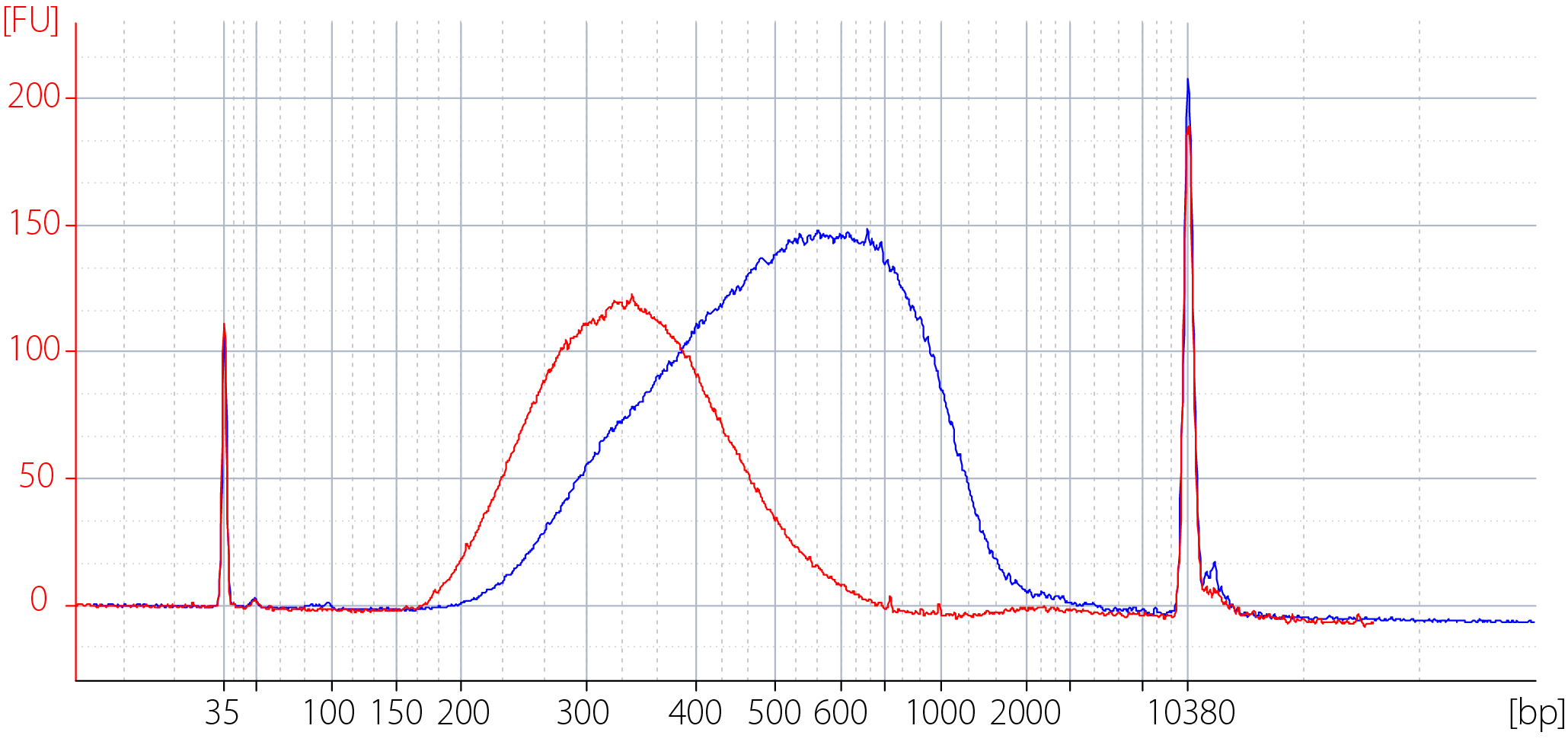
使用する逆転写試薬に応じて、2種類のプロトコル(Short Size Inserts (RTM)プロトコルとLong Size Inserts (RTL)プロトコル)が用意されていることにご注意ください。 PCRシステムが更新され、PCRがPMに、E3がPEに置き換えられました。これに伴いPCRプログラムもアップデートされました(下記参照)。
以下がプロトコルです。変更点は緑色のフォントで表されています
逆転写
- Prepare a mastermix of 14 µl RTM or RTL and 1 µl DSP per sample. Mix well.
- Add 15 µl of RTM or RTL / DSP mastermix to 10 µl RNA sample. Mix well.
- Incubate for 3 min at 94 °C, then 15 min at 16 °C for RTM.
- Incubate for 1 min at 94 °C, then 15 min at 16 °C for RTL
- Prepare a mastermix of 4 µl RTM or RTL and 1 µl E1 per sample. Mix well.
- Add 5 µl RTM or RTL / E1 and mix well.
- Incubate: 10 min at 25 °C, 40 min at 37 °C, 10 min at 42 °C, cool to 25 °C. Proceed immediately to purification!
エンドポイントPCR
- Prepare a mastermix with 7 UL PM and 1 UL PE per reaction. Mix well.
- Add 8 UL of PM / PE mastermix to 17 μl of each purified library.
- Add 10 µl of one Unique Dual Index Primer pair (UDI12A_0001-0384, or UDI12B_0001-0096) to each sample.
- Conduct 11 - 25 cycles of PCR (as determined by qPCR assay) with the following program:
- 95 °C for 15 seconds,
- 60 °C for 15 seconds, and
- 72 °C for 60 seconds,
いいえ、CORALL V2 は、CORALL V1と同様に優れた性能と再現性を備えています。
CORALL V2は、Unique Molecular Identifiers (UMI)を内蔵しているため精度が向上しており、ライブラリ作製が困難なサンプルでも優れた性能を発揮します。
また、CORALL V2キットにはUnique Dual Indexes (UDI)が含まれており、利用することですべてのシーケンサーでシーケンス出力を最大化することができます。
CORALLはtotal RNAをinputとして使用できますが、マップピングされたシーケンスリードの90%以上を占めるリボソームRNAを除外するために、CORALLライブラリ調製の前にリボソームRNAの除去またはPoly(A)RNAのセレクションを行うことを推奨しています。
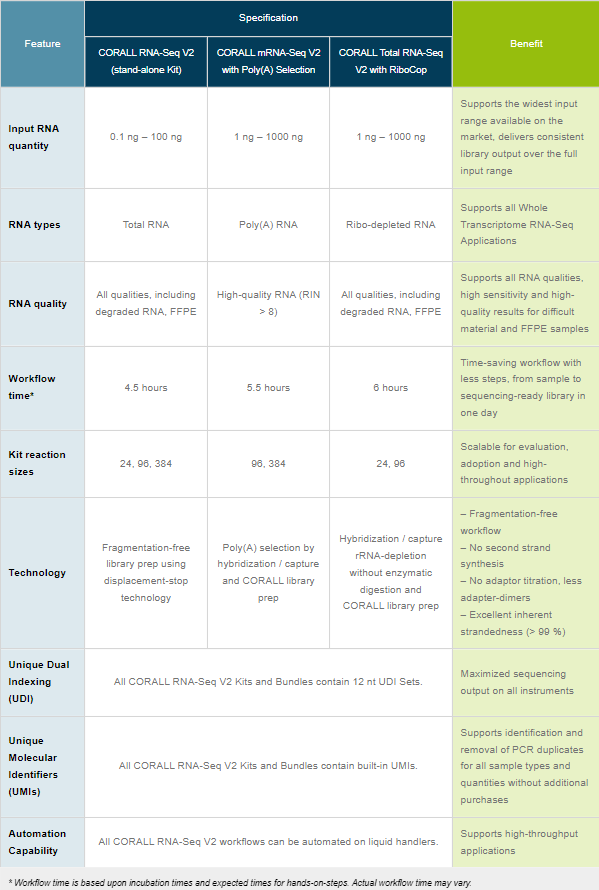
CORALL RNA-Seq kit(品番:171 - 176)
リボソームRNAを除外キットやPoly(A)RNAのセレクションキットは含まれません。
CORALL mRNA-Seq Kit (品番:177 - 182)
Poly(A)RNAのセレクションを行うことができる Poly(A) RNA Selection Kit が含まれています。
ポリアデニル化mRNAを産生するすべての生物種に適しています。このキットは、ポリアデニル化済みのサンプルのライブラリ調製にも使用できます。
Poly(A) RNA Selection Kit (品番:157) は別途購入することもできます。
CORALL Total RNA-Seq kit (品番:183 - 186)
ヒト、マウス、ラットのリボソームRNA除去用のRiboCop kit(品番:144)が含まれています。品番:184-186にはRiboCop kitに加えて、グロビン除去用の試薬が含まれます。
RiboCop kitは、ヒト、マウス、ラット用(品番:144、またはグロビン除去試薬付きのCat. No. 145)、バクテリア用(品番:125 - 127)、酵母用(品番:190)があり、個別に購入できます。
全てのCORALL RNA-Seq kitに12 nt Unique Dual Indexing (UDI) Setが付属します。どのキットも12 nt Unique Dual Indexing Solutions (UDI)の使用を推奨しております。12 nt UDI Sets詳細については、12 nt UDI 製品ページをご覧いただくか、 12 nt UDIFAQ をご覧ください。
付属品の詳細は以下をご確認ください。
CORALLは幅広い生物種に対応しており、十分なinputがある限り、由来に関係なく(バクテリアを含む)すべてのRNAに対してご使用できます。正しいデータ解析を行うためには、シーケンスリードを比較するための適切なリファレンスゲノムが必要です。
CORALLはtotal RNAをinputとして使用できますが、マップピングされたシーケンスリードの90%以上を占めるリボソームRNAを除外するために、CORALLライブラリ調製の前にリボソームRNAの除去またはPoly(A)RNAのセレクションを行うことを推奨しています。
CORALL RTMとRTLの主な違いは逆転写試薬であり、最終的なライブラリのインサートのサイズに影響を与えます。
RTM試薬
- CORALL V1標準プロトコルと同等であるため、発現プロファイリングや、FFPEサンプルなど低品質RNAの処理に最適です。
- 期待されるライブラリの平均サイズは約350 bpです。
- 時間とコストを節約しながら、発現プロファイリング(推奨長: SR50 - 150)のためのシングルリードシーケンシングを可能にします。また、ペアエンドシーケンシング(推奨長: PE50-75)にも対応しています。
RTL試薬
- より長いリードを得るために最適化されており、選択的スプライシング、融合トランスクリプトの検出、アイソフォーム同定、トランスクリプトのアノテーション(de novoアセンブリーを含む)などのアプリケーションに適しています。
- 期待されるライブラリの平均サイズは約550 bpです。
- ペアエンドシーケンス(推奨長:PE100〜150)を必要とする困難なアプリケーションに最適ですが、シングルリードモード(推奨長:SR150)にも対応しています。
はい、CORALL RTMとRTLで作成されたデータは高い相関性を示しています。ただし、インサートサイズの違いにより、いくつかの違いが検出される可能性もあります。
分解した RNA および FFPE サンプルを RTL で処理することはお勧めできず、RTMをご使用ください。RTLはより長いインサートを生成するように設計されているため、インタクトなinput RNAで使用する必要があります。分解した/FFPE RNAでRTLを使用した場合、ライブラリの長さが短くなることで収量に影響が出たり、アーチファクトが多く発生する可能性があります。
ライブラリ増幅に最適なPCRサイクル数を決定するには、qPCRアッセイを推奨します。これにより、サイクル数が不足したり過剰になったりするのを防ぎます。サイクル数が不足すると、シーケンシングに十分なライブラリが得られない可能性があり、逆に過剰になると重複率が高まり、定量性を損なう可能性があります。
qPCRアッセイは、RNAの初期量が少ないサンプル、FFPEサンプル、低品質サンプルや品質が変動するサンプルに対して特に重要です。
また、異なるサンプルタイプ(種、組織、細胞タイプ)や異なるRNA処理ステップ(rRNA除去やpolyA selectionなど)の使用はサンプルのRNA含有量に影響を与えます。そのため、サンプルタイプ間でinput量が一致していても必要なPCRサイクル数が異なる場合があります。
qPCRアッセイを行うには、CORALL Total RNA-Seq Library Prep Kitと一緒に以下の試薬をご購入ください:
- PCR Add-on and Reamplification Kit V2 for Illumina (Lexogen, 品番:208)
- SYBR Green I Nucleic Acid Stain, 10,000x in DMSO(Sigma, 品番:S9430または ThermoFisher , 品番:S7585)
注意事項
SYBR Greenを含むqPCRマスターミックスやEvaGrenなどの他のフルオロフォアの使用は推奨されません。これらの使用はパフォーマンスが異なり、エンドポイントPCRの必要サイクル数の計算に影響を及ぼす可能性があります。他のフルオロフォアを使用した場合、増幅曲線が遅延し、最大蛍光強度が低下することがあります。そのため、エンドポイントサイクル数は異なる計算が必要であり、エンドポイントPCRテストで確認する必要があります。
パッシブリファレンス染料(例えばROX)は必要なく、qPCRマスターミックスには含めないようにしてください。
qPCRアッセイの詳細についてはFAQをご覧ください。
qPCRアッセイからエンドポイントPCRサイクル数を計算する方法についてはFAQをご覧ください。
※特定のサンプルタイプや入力量でCORALL V1でエンドポイントPCRのオプションサイクル数を以前に決定した場合、CORALL V2でも同じサイクル数を使用できます。
CORALL ライブラリは、シングルリード (SR) モードまたはペアエンド (PE) モードでシーケンスできます。
- RTM で生成されたライブラリの場合は、SR50〜150 および PE50〜75 を推奨します。
- RTL で生成されたライブラリの場合は、SR150 および PE100〜150 を推奨します。 シーケンス形式の選択は、特定のアプリケーションによっても異なる場合があります。
こちらをご参照ください。
Lexogenが提供する解析サービスであるKangooroooをご利用ください。キットに付属するvoucher codeを利用すれば、無料でRNA-Seq データを解析することができます。
ファイルサイズについて
voucher codeを利用しての無料データ解析のFASTQ入力ファイルのサイズ制限は5 GBです。これより大きなFASTQファイルサイズは、別途費用がかりますのでお問い合わせください。
ご自身でデータ解析パイプラインを設定したい場合は、GitHubに公開されているCORALLパイプラインをご覧ください: GitHub - Lexogen-Tools/corall_analysis: Standard analysis routine for samples generated with Corall and Corall v2 library preparation kit
CORALL RNA-Seqデータは、ほとんどの標準的なRNA-Seqパイプラインを使用して解析することも可能です。目的に応じて、ワークフローの調整が必要になる場合があります。
CORALLデータ解析に関わる基本的な手順と推奨ツールの概要は以下をご覧ください
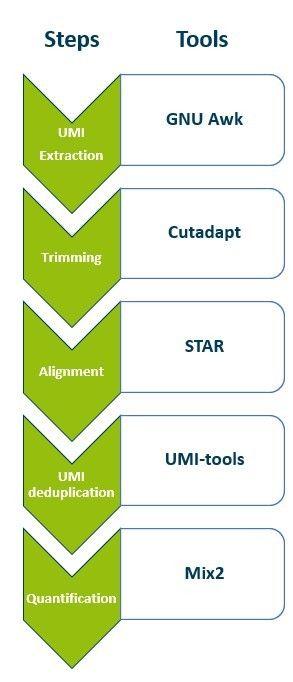
図. CORALLデータ分析パイプラインのワークフロー
以下のLexogen Tools Github ページから CORALL データ分析パイプラインをこちらからダウンロードできます。
このパイプラインは、CORALL mRNA-Seq および Total RNA-Seqデータでの使用に互換性があります。
通常、FFPE や分解 RNA のinputに CORALL mRNA-Seq を使用することはお勧めしません。Poly(A)セレクションはoligo (dT) bead captureによって行われるため、分解RNAをinputとして使用した場合、転写産物の大部分で5'領域を補足できないことになります。転写産物の長さはリードカウントを正規化するための基準として使用されるため、正確な転写産物量の推定に悪影響を及ぼします。 RNA integrityまたはquality scores(RIN、RQN)が 8 未満のサンプルについては、CORALL ライブラリ調製の前にリボソーム RNA除去(Lexogen社のRiboCop rRNA Depletion Kitsの使用など)を行うことを強く推奨します。
さらに、RTLは分解またはFFPE処理したRNAサンプルでの使用は推奨されません。RTLが正しく機能するためには、完全なinput RNAが必要です。
poly(A)セレクションを行ったTotal RNAのinput量は1 ng 〜 1 ugです。 CORALL mRNA-Seqを使用する場合、Poly (A) 選択のinputとして高品質 RNA (RIN >8) が必要です。
CORALL Total RNA-Seqで使用できます。ただし、RTLは分解/FFPE RNAサンプルには推奨しません。分解/FFPE RNAサンプルにはRTMを使用してください。
分解したRNAおよびFFPEサンプルからのライブラリ調製をCORALLライブラリ調製キットで行う場合、プロトコルの修正が推奨されます。
以下の推奨事項は、total RNA input 25〜500 ngで評価されています:
- 中間クリーンアップステップを行わずに、rRNA除去前にDNase I処理を行います。
- リボソームRNA除去を行います(例:LexogenのRiboCop rRNA Depletion Kitsを使用)。
- CORALL V2ライブラリ調製プロトコルのステップ33で、31.5 ULではなく42 ULのPBを使用します。
- LexogenのSpike-In RNA Variant controls(SIRVs)を使用する場合、これらはDNase I処理前にTotal RNAにスパイクインする必要があります。推奨されるスパイクイン量は、シーケンスリードの0.1〜0.2%がSIRVsにマップされるように減らすべきです(詳細はSIRV User GuidesおよびHow to use SIRVs FAQsで説明されています)。SIRVスパイクイン計算ワークシートはSIRVsダウンロードページから入手できます。
最小シーケンスリード長は 75 bp を推奨しています。シングルエンドまたはペアエンドのシーケンス形式を使用できます。
CORALL Total RNA-Seq は、total RNAをinputとして使用するか、リボソームRNA除去を行った後に実行されます。
リボソームRNAを除去した場合は、1 ng 〜 1 ug のTotal RNAをinputします。rRNA 除去には LexogenのRiboCop rRNA Depletion Kitsのご使用をお勧めします。CORALL の上流で他のリボソームRNA除去キットを使用する場合は、CORALL ユーザーガイドに記載されている各 rRNA除去キットのインプット仕様に従ってください。
リボソームRNAを除去やPoly(A)濃縮を行っていないTotal RNAの場合は100 pg 〜 100 ng を推奨しております。
