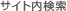ゲノム編集は、まず標的DNAに効率よく二本鎖切断(DSBs)を導入することから始まります(図.1)。二本鎖切断(DSBs)は相同性組換え(HR)あるいは、相同性修復テンプレートが存在しない場合において、非相同性末端結合(NHEJ)を介して修復されます。NHEJは破損末端に短鎖の挿入または欠損を導入しこれらを結合します。この挿入欠損を生ずる傾向を簡便な遺伝子ノックアウト手法として利用します。FokIヌクレアーゼと融合した1対のDNA結合タンパク質からなるTALENと、Cas9ヌクレアーゼと標的特異的一本鎖ガイドRNA(sgRNAs, guide RNA, gRNA)との複合体であるCRISPR-Casの双方とも、HRまたはNHEJを介してDNAを編集します。
図.1 ゲノム編集ツールにより誘発された二本鎖切断(DSBs)の修復経路
左:非相同性末端結合
右:ドナーテンプレート存在下での相同組換え
CRISPR-Cas と TALENのどちらを利用するかを検討する際に考慮する4つのポイントを以下に示します。
【1】 特異性
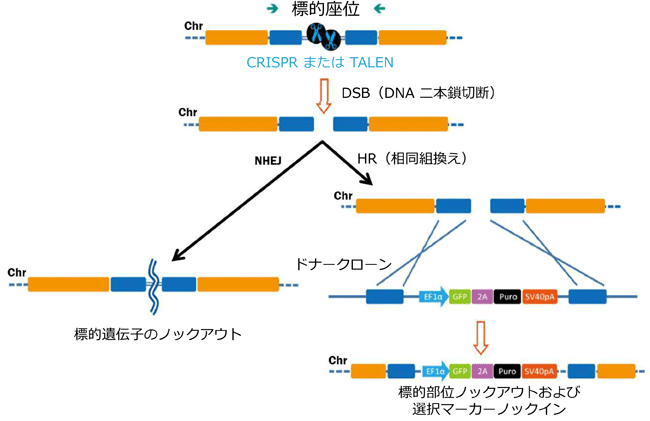
CRISPR-Cas と TALENの何れの手法も非常に高い標的特異性をもち、正確な遺伝子変異を導入することができます。CRISPR-Casの場合、2種の天然に存在する短鎖RNAを人工的に融合させたsgRNAを介してこの特異性を獲得します(Jinek, et al. 2012)。sgRNAは、S. pyogenes 由来のCas9ヌクレアーゼを染色体上の20塩基の標的部位に配向させますが、この標的配列はPAM(スペーサー前隣接モチーフ)といわれるN-G-Gの3塩基が直後に続く必要があります(図.2)。sgRNAはPAM部位の反対鎖とハイブリダイズしCas9ヌクレアーゼがDNAを切断します。近年、数々の論文においてCRISPR-Casは高頻度で標的部位に二本鎖切断(DSBs)を形成することが示されています。しかし、sgRNAは様々な度合いで許容性を示し、期待しない部位を最大5ヶのミスマッチ(ワトソン - クリック塩基対ではない)をもって標的する可能性があり(Fu, et al., 2013)、そのため、CRISPR-Casはゲノム上の数々のオフターゲット部位と物理的に会合することも示されています(Kuscu, et al, 2014)。高頻度で意図しない部位に挿入欠損を形成することがいくつか報告されている(Fu, et al. 2013)ものの、他の研究者からはオフターゲット変異誘発性に関する当初の見積もりが誇張されていたことも指摘されています(Li, et al., 2013; Yang, et al., 2013; Wang, et al., 2014)。
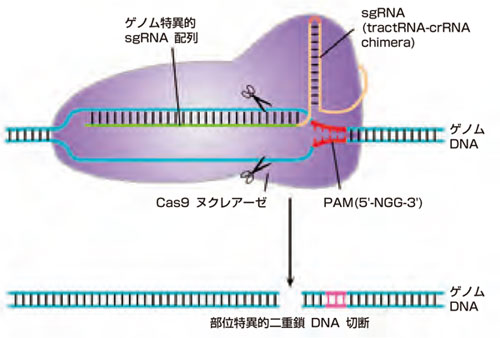
図.2 CRISPR-Cas9標的認識
近年の技術開発によりCRISPR-Casの特異性は飛躍的に改善しています。ひとつの方策は、標的部位を挟んだ両鎖を2種類のsgRNAで標的し、1対の一本鎖切断(“ニッカーゼ”)変異を利用するものです(Mali, et al., 2013; Ran, et al., 2013)。この場合であっても各ニッカーゼがオフターゲットを生ずるものの、ニックはDSBに比べて変異原性が低いため、1対のニッカーゼを利用することでオフターゲットを劇的に低減できます。さらに、sgRNAを最短17塩基まで短くすることでオフターゲット効果を低減することもできます(Fu, et al., 2014)。不活性型Cas9とFoklエンドヌクレアーゼ(RNA誘導性FoklヌクレアーゼまたはRFNs:Tsai, et al., 2014)の融合にはオフセットsgRNAペアに媒介される二量体形成が必要であることから、この方法では一対のニッカーゼや短鎖sgRNAを利用するよりもさらにオフターゲット変異誘発性を軽減することができます。
一方、TALENではオフターゲットの問題は低いといわれています。一般に、TALENは34アミノ酸の18回繰返しで構成されます。この繰返しは“反復可変二残基”(RVD)とよばれ、アミノ酸第12位と13位において可変です。RVDを介するDNA結合コード(図.3)によりDNA結合特異性が得られます。TALENペアは標的部位において、14〜20塩基からなる“スペーサー”によって分割された反対鎖にそれぞれ結合する必要があります(図.4)。Foklが活性を示すためには二量体を形成する必要があるため、このオフセットデザインが必要となります。ゲノム上では、このように長鎖のDNA結合部位(約36塩基)は非常にまれであると考えられます。RVD-DNA結合コードには縮重がみられることもあり(Bogdanove and Voytas, 2011)、TALENにおけるミスマッチ寛容やオフターゲット活性はほとんど報告されていません。近年のiPS細胞株を高活性TALENにより編集した例では、標的部位と相同性をもつ他のゲノム領域において変異原性活性がみられなかったと報告されています(Park, et al., 2014)。
図.3 TALEN用DNA結合コード。Bogdanove & Voytas (2011)より引用
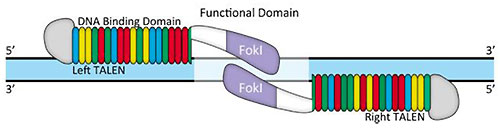
図.4 一般的なTALENデザイン
【2】 標的部位選択
上述の通り、CRISPR-CasのsgRNA標的はNGG部位(PAM配列)の直前に位置する必要があります。ノックアウトを目的とする場合、GG部位を探すことはそれほど困難ではありませんが、他のアプリケーションでは問題となることもあります。さらに、ダブルニッキング法とRFN手法を利用する場合、標的デザインに制約がかかります。何れの場合も2つのガイドRNAはPAM部位がお互いに末端に位置する(尾部と尾部に配向:Mali, et al. 2013; Ran, et al., 2013; Shen, et al. 2014; Tsai, et al., 2014)よう方向付けを行う必要があります。各sgRNA尾部の間隔はCRISPR-Casのデザインに影響する決定的な要因となります。一対のニッカーゼは-8〜30塩基のオフセットをもつときに最もよく機能します(最適な間隔は0〜20塩基)。RFNでは、その間隔はより限定的です。13〜18塩基の間隔が必要であり、最適な間隔は16〜17塩基となります。PAM部位要求性と併せると、アプリケーションによってはペアードCRISPR手法の利用が困難となる場合があります。一方で、TALENデザインでは決められた間隔をもつオフセット結合タンパク質が必要となるものの、デザイン上の制約は現時点でこれ以外に報告されていません。従って、TALENペアによりゲノム上の如何なる部位も標的でき、標的部位選択においてCRISPRに比べてより自由度と柔軟性が高いともいえます。
【3】 ゲノム編集効率
CRISPR-Casは染色体上の標的を高頻度で修正可能であることもあり、多くの研究者に人気です。挿入欠損形成率が70%以上であるとの報告もあります。予想されたように、切断型sgRNA、1対のニッカーゼ、およびRFNでは挿入欠損形成効率がより低くなります。
TALENもまた、高い効率で染色体修正が可能です。例示(Park, et al., 2014)した通り、1つのTALENペアでは挿入欠損形成率が33%でした。この挿入欠損産生効率は全てのCRISPR-Cas法に匹敵するものです。しかし、TALENはシトシンメチル化に感受性であり、特にCpGジヌクレオチドにおいて感受性が高いことが知られています。これはDNAサイレンシングにおいて一般的かつよく知られた機序です。CpGメチル化はプロモータ領域において最も散見されますが、タンパク質をコードするDNA配列においても生じます。TALENペアによっては変異誘発活性がほとんどない、または全く示さないものがありますが、このメチル化への感受性が要因ではないかと考えられています。
【4】 デザインと構築の簡便性
CRISPR-Casはデザインやその使用が簡便です。標的部位ごとに必要な作業は、「20塩基のゲノム標的部位をsgRNAに組込むこと」のみです。プラスミド構築は簡単かつ単純であるため、複数の標的遺伝子を扱うことも容易です。実際のゲノム編集実験では、sgRNAは再利用可能なCas9ヌクレアーゼと同時発現します。ダブルニッキング法やRFN手法では、一対のsgRNAが協調して機能するよう選択してデザインする必要があるものの、sgRNAデザインや構築は上記と同様です。第一次世代のCRISPR-Casでは標的デザインはほぼいつでも成功するとの評判でしたが、切断型sgRNA、ダブルニッキング法、またはRFNではこの限りではありません。論文報告に準じてデザインした場合であっても、デザインによっては成功することもありますが、うまくいかないものもあります。また、CRISPRはメチル化に対して感受性ではありません。
TALENを構築する場合、標的ごとに新規タンパク質を再設計する必要がありますが、TALENデザインはクローニング量が削減可能な反復併用型モジュールの利用によって合理化されてきています。上述の通り、TALENはシスチンメチル化に感受性があり、5-メチルシトシンにはN* RVDを利用する方法もあります(図.3)が、標的部位がメチル化されていることを認識しておく必要があります。本課題の対処法のひとつとしてCpG部位を避けることが挙げられます。もちろん、これにより新たな制約を与えることになるものの、もし成功すればゲノム編集ツールとしてTALENがより魅力的なツールとなるでしょう。
CRISPR-Casはより新しく簡便で複数遺伝子の編集が可能な技術です。TALENはその比較的制約の少ない標的部位要求性や高い特異性から未だに利用価値の高い手法であるといえます。TALENとCRISPR-Casの何れを使用するべきか決める際には、これら2つのシステムの違いをよく理解しておくことが重要です(表.1)。実験計画におけるアプリケーションの種類も考慮する必要があります。例えば、試験的な実験の一環として遺伝子ノックアウトを迅速に行いたい場合、第一次世代のCRISPR-Casが妥当ではないでしょうか。比較的制約の少ないデザインでオフターゲットの少ない変異誘発を行う必要がある場合には、TALENを試すことも有用です。
表.1 TALENとCRISPR間の比較
| 特性 |
TALEN |
CRISPR-Cas9 |
| 認識の種類 |
タンパク質-DNA |
RNA-DNA |
| 複数遺伝子の標的 |
○ |
◎ |
| メチル化 |
感受性 |
感受性ではない |
| オフターゲット効果 |
確認されたオフターゲット効果が少ない |
TALENやZFNに比べよりオフターゲット効果の可能性が高い |
まずはできるだけ安く、簡単にはじめたい!
Human,Mouse,Rat遺伝子を標的するgRNAをすべてデザイン済!プラスミド、レンチウイルス粒子、アデノウイルス粒子のなかからお好みのフォーマットをご利用頂けます。Casタンパク質は、野生型Cas9もしくはニッカーゼタイプから選択し、Co-transfectionして使用します。同じ研究室内で複数の細胞を扱う場合にはこちらがおすすめです。
絶対に、確実に、標的遺伝子をノックアウトしたい!
Human,Mouse遺伝子を標的するgRNAをすべてデザイン済で、確実なノックアウトを行えるよう、gRNAと野生型Cas9を同時に発現するAll-in-Oneタイプの発現コンストラクトが、1遺伝子に対して2種類、さらに、二本鎖切断(DSBs)が導入された細胞のみを選択するためのドナーベクター(GFP-Puro 挿入用)、スクランブルコントロールベクターが1つのキットになった便利な商品です。
いくつも標的遺伝子があるので、ご自身でgRNAを挿入したい!
CRISPR-Casシステムの最も素晴らしい点のひとつは、gRNAを変更するだけで標的遺伝子をいかようにも変更できる点です。複数遺伝子を同時にノックアウトすることも可能です。複数の標的遺伝子があり、そのたびにコンストラクトを購入するのは大変…という場合にはこちらがおすすめです。
実績のあるgRNA配列を利用したい!
サンタクルズ社では、3種のgRNA+Cas9発現プラスミド(非ウイルス型/All-in-Oneタイプ)を混合したプール型のCRISPR-Cas9 ゲノム編集ツールをご提供しています。各gRNA配列は、GeCKO(Genome-scale CRISPR Knock-Out)ライブラリーと同一です。HumanおよびMouse用のgRNA+Cas9発現プラスミドをゲノムワイドにご用意しています。
ノックアウト細胞作製を受託したい!
- Applied Biological Materials(APB)社 iCRISPR ノックアウト細胞作製受託サービス
Applied Biological Materials (アプライドバイオロジカルマテリアルズ/APB) 社は、全ヒト、マウス、およびラット遺伝子を標的とした独自のゲノムワイド sgRNA ライブラリー発現用のレンチウイルスシステムを開発しました。本サービスでは、これを用いてヒト、マウス、ラット由来細胞の標的遺伝子をノックアウトします。厳格な品質管理とサンガーシーケンス、PCR 又はウエスタンブロットによる遺伝子ノックアウトの検証後、ノックアウト細胞をお客様にお届け致します。