GeneCopoeia(GCP)社のCRISPR-Cas9用非ウイルス性プラスミドは一般的な手法でトランスフェクションできますが、特定の細胞タイプに最適化および検証済のEndoFectin™ トランスフェクション試薬もご提供しています。使用細胞に適した手法・試薬をご選択ください。GeneCopoeia(GCP)社のCRISPR-Cas9レンチウイルスプラスミドは、第3世代のレンチウイルスパッケージング試薬に対応しており、Lentifect™ レンチウイルスパッケージング試薬やレンチウイルスパッケージングサービスもご提供しています。非ウイルスプラスミドとレンチウイルスプラスミドのいずれも、トランスフェクションやトランスダクションの作業工程が簡単になるよう、薬物選択マーカーと蛍光レポーター遺伝子を保有しています(図.3)。
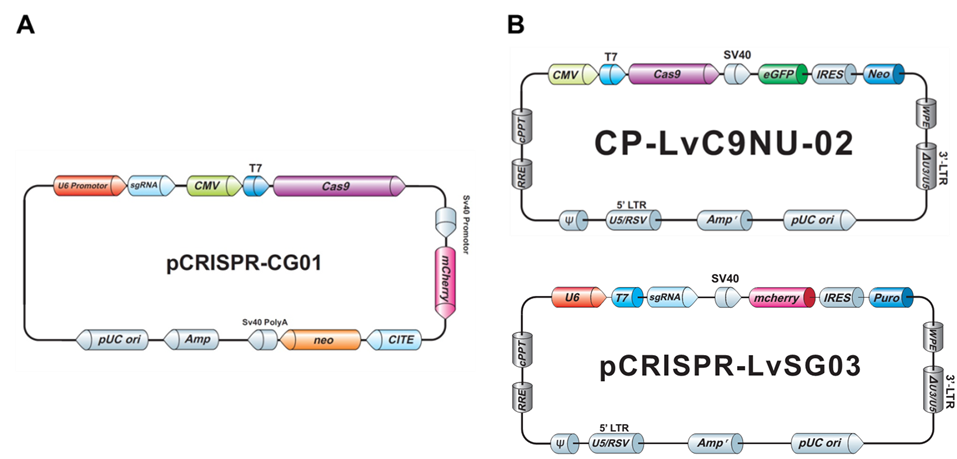
図.3 GeneCopoeia(GCP)社ゲノム編集プラスミドの一例
A. CRISPR Cas9ヌクレアーゼ、1種類のsgRNA、G418選択用neo遺伝子、および蛍光レポーターとしてmCherryをコードした非ウイルス性のall-in-oneプラスミド
B.上部:neo遺伝子とeGFPをコードしたCRISPR Cas9ヌクレアーゼベクター(レンチウイルスプラスミド)
下部:ピューロマイシン選択用puro遺伝子とmCherryをコードしたCRISPR sgRNAプラスミド(レンチウイルスプラスミド)
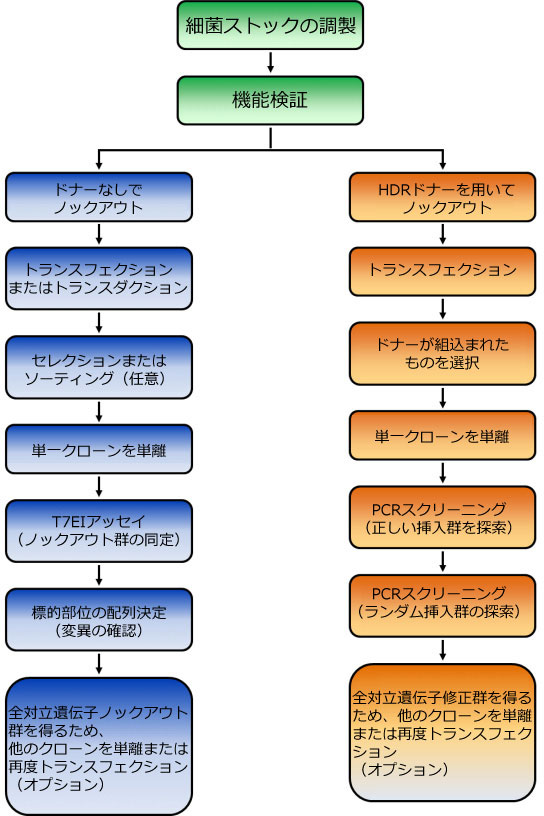
遺伝学では、「クローン単離」として知られるように、集団から離れた変異体を精製することが重要です。クローン単離を行うことで、結果の解釈を混乱させかねない細胞分裂やオフターゲット作用により生ずる期待しない修正による影響を最低限に抑えることができます。スクリーニングを始める際は、まず、単一コロニーを形成できるよう細胞を播種してディッシュから取り出すか、96穴プレートで段階希釈をとります。
実施するスクリーニングのタイプは、どのようなゲノム編集を行うかによって異なります。ここでは、3種類の一般的な状況を論議します。
■ NHEJ(非相同末端結合)によるノックアウト
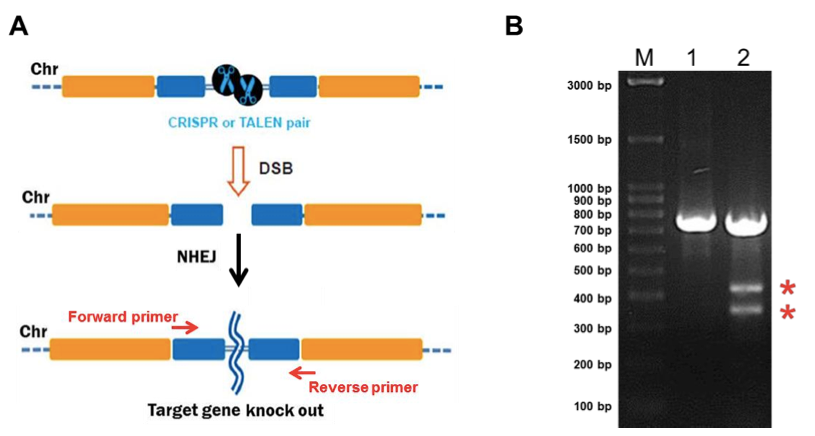
NHEJ(非相同末端結合)介在性ノックアウトを迅速にスクリーニングする方法として、機能検証の項に記載のあるミスマッチ切断アッセイがあります。修正されたクローンが同定できた後は、PCR産物を用いてサンガーシークエンス法による配列決定を行い、変異状態を同定する必要があります。GeneCopoeia(GCP)社では、クローンスクリーニングに利用できるミスマッチ切断アッセイキットをご提供しています。
■ ドナープラスミドを用いたHR(HDR)アプリケーション
一般に、TALENやCRISPR-Cas9を介する相同性組換え頻度は、NHEJ(非相同末端結合)と比べてかなり低いため、ドナープラスミドにより修正されたコロニーを同定するにはより多くのコロニーをスクリーニングする必要があるように思われるかもしれません。しかし、ドナープラスミドにより薬剤選択や、蛍光レポーターを導入することができます。GeneCopoeia(GCP)社のドナープラスミドは、薬剤選択や蛍光レポーター遺伝子をもっています(図.4)。トランスフェクション後、ピューロマイシン等の薬剤存在下において細胞を低密度で播種し、単一コロニー形成を促します。あるいは、96ウェルプレートを用いて段階希釈をとる方法もあります。薬剤存在下で生長するコロニーが、修正部分を保持する安定細胞株構築候補です。
薬剤耐性や蛍光レポーター発現クローンが同定できると、その後の手順はNHEJ(非相同末端結合)を利用する場合と劇的に異なります。ノックアウトの場合、通常、セレクション/レポーターカセットがタンパク質コード領域に組込まれ、CRISPR-Cas9により切断された部分に短鎖欠損をもつことも時々あります。その他、DNAの特定染色体領域が削除されカセットと置換される場合や遺伝子がインフレームの融合タグを保有する場合もあります。スクリーニングを行う場合、未修正の染色体配列と挿入配列との境界部分にまたがるプライマーを利用してPCRを行います。挿入配列の左と右側の両方において、マーカーと同方向に合成するよう相同腕用に1つのプライマーを設計し、反対側のプライマーは相同腕に向かって合成するよう挿入配列に設計します。もし組込みが良好に行われた場合には、アガロースゲル上で期待される大きさのバンドを確認することができます。
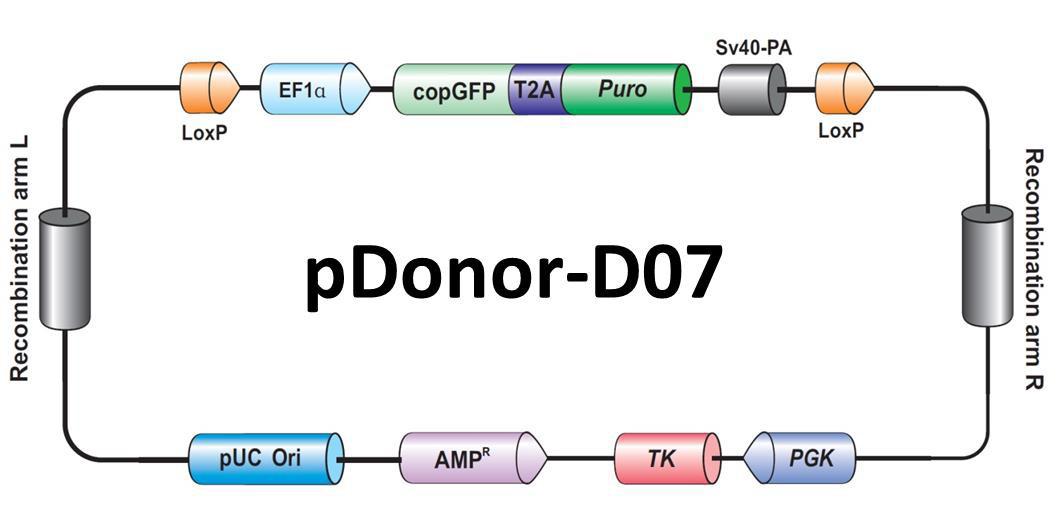
図.4 HR(HDR)アプリケーション用GeneCopoeia(GCP)社ドナープラスミドの一例
薬剤選択を行うとドナープラスミドのランダム組込みを招く恐れがあり、除外する必要があります。GeneCopoeia(GCP)社のドナーベクターは組換え領域の外側に単純ヘルペスウイルス(HSV)チミジンキナーゼ(TK)遺伝子をもっています(図.4)。ガンシクロビルを添加した選択培地を使用すると、ドナープラスミドがランダムに組込まれた細胞は通常死滅するため、正常なドナー組込みが生じた細胞群を濃縮することができます。それほど頻繁ではありませんが、ランダム組込みによりTK遺伝子が排除されることもあるため、PCRやサザンブロットを行ってこれらを除去するほうがよいでしょう。
最後に、HR(HDR)により修正された遺伝子は、修正部位周辺の染色体構造を確認し、導入した変異が存在することを検出するため、該当する場合には配列決定する必要があります。 哺乳動物細胞システムにおけるCRISPR-Cas9のデザイン、構築およびその利用に関する専門知識や経験に富むGeneCopoeia(GCP)社では、sgRNAやHDRドナーデザインから作業を開始し、配列検証済みプラスミドをご提供しています。また、CRISPR-Cas9構築や機能検証などのカスタムサービスもご提供しています。
トランスフェクションやスクリーニング計画を考える前に、スクリーニングするコロニー数に影響を及ぼす可能性がある複数の要因や、必要な場合には繰返し実施するトランスフェクションの回数に関して熟考する必要があります。
■ トランスフェクション効率
CRISPR-Cas9 ゲノム編集ツールを受け取るであろう細胞の割合は、スクリーニングを行うコロニー数を決定する上で非常に重要です。もし使用細胞株におけるトランスフェクション効率が40%の場合、トランスフェクション効率が80%の細胞に比べて2倍もの数のコロニーをスクリーニングすることが予想されます。もし使用細胞株のトランスフェクション効率が低い場合、プラスミドにコードされた蛍光マーカーを使ってソーティングしてトランスフェクションされた細胞を濃縮したり、短期的には薬剤選択を利用することもできます。GFPを始めとする蛍光レポーターを定常発現するプラスミドを利用して使用細胞株のトランスフェクション効率を推定することを推奨します。
■ コピー数
完全なノックアウトや変異誘発性を得るためには、標的遺伝子の全ての対立遺伝子を修正する必要があるかもしれません。二倍体細胞株を使用する場合、2つの対立遺伝子を修正する必要性があると想定する研究者が多くいます。しかし、HeLaを始めとする一般的な不死化細胞株の多くは、異数体または倍数体であるため、対象遺伝子を2コピー以上保有している可能性があります。1つの細胞内では、複数の対立遺伝子修正に比べて単一対立遺伝子修正効率の方が高くなります。したがって、もし2つ以上の対立遺伝子を修正する必要がある場合には、2対立遺伝子修正に比べてより多くのコロニーをスクリーニングする必要があると考えられますので、複数回にわたってトランスフェクションを行う必要があるかもしれません。
■ 切断効率
CRISPR-Cas9による挿入欠損形成効率は、通常、約5%〜70%です。トランスフェクション効率の場合と同様に、切断効率が40%の場合に比べ、それが20%の場合は2倍ほどの数のコロニースクリーニングが予測されます。
修正クローンを同定するためのスクリーニングに使用するべきコロニー数は、使用する細胞由来の要因や、要因の組合せに依存します。単純計算すると、もし50%の切断効率をもつCRISPR-Cas9 ゲノム編集ツールを用いてトランスフェクション効率が60%の二倍体細胞株の2対立遺伝子ノックアウトを得たい場合、単一対立遺伝子が修正されたコロニーの割合は0.5 × 0.6 = 30%となります。2対立遺伝子修正の場合、陽性コロニーの割合は15%となります。以上を考慮すると、2対立遺伝子修正をもつクローンを1つ同定するためには少なくとも20のコロニースクリーニングが必要となります。もし、トランスフェクションと切断効率がより低い場合で、使用細胞株に標的遺伝子が2コピー以上存在する場合、わずか1つの完全なノックアウト細胞を単離するために、より多くのコロニースクリーニングが必要となります。