





















セルバイオラボ社 アデノウイルスによる遺伝子デリバリー プロトコール内で使用した商品

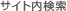
セルバイオラボ社 アデノウイルスによる遺伝子デリバリー プロトコール内で使用した商品
I:miRNA Precursor のクローニング
mature miRNA の過剰発現用にmiRNA precursor を哺乳動物発現ベクター(pacAd ベクター、図2) にクローニングします。
・ miRNA precursor クローンのデザイン例(ヒトlet-7a-2 miRNA の場合)
1.miRNA のステムループ配列を Sanger' s miRNA database からダウンロードします。
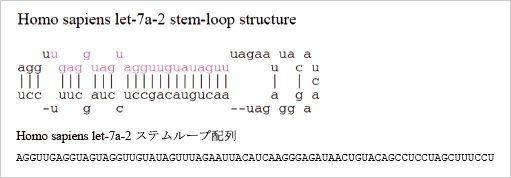
2.miRNA ステムループ配列をBlast で検索します。
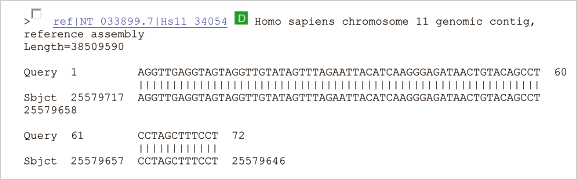
3.PCR とクローニング
1) 100bp のネイティブフランク配列をmiRNA の上流と下流に加えます。(100bp のflank 配列を含むHuman let - 7a - 2 miRNA precursar 配列の例)
GCCCAAATAGGTGACAGCACGATGAATCATTATAAGACTAACTTGTAATTTCCCTGCTTAAGAAATGGTAGTTTTCCAGCCATTGTGACTGCATGCTCCCAGGTTGAGGTAGTAGGTTGTATAGTTTGAATTACATCAAGGGAGATAACTGTACAGCCTCCTAGCTTTCCTTGGGTCTTGCACTAAACAACATGGTGAGAACGATCATGATTCCTCCAGGCCTTTTCTCCCTATGAAAGGTAAGATTGGGTACGTTATTTTATGGTATTT
2) BamHI サイトを含むフォワードプライマー,NheI サイトを含むリバースプライマーを設計します。
(例)ヒトlet-7a-2 miRNA precursor の場合
Forward PCR Primer: tcga-ggatcc (BamHI)-21 nt
Reverse PCR Primer: tcga-gctagc (NheI)-21 nt
3) ゲノムDNA 由来のmiRNA をPCR で増幅させ,発現ベクターのBamHI/NheI サイトにそれぞれクローニングします。
4) インサートをDNA シーケンスによって確認します。
Forward Sequencing Primer: TTTGCACCATTCTAAAGAAT
Reverse Sequencing Primer: AAACCTCTTACATCAGTTAC
II:リコンビナントアデノウイルスの調製
1.ベクターをPac I によってリニア化します。
1) 十分量の目的遺伝子のクローニングされたpacAd5 シャトルベクターとpacAd5 9.2-100 Ad バックボーンベクターをPac I で制限酵素処理します。
2) 制限酵素処理したそれぞれのDNA と処理していないDNA 0.5μg を0.8%アガロースゲルに流し,完全にPacI で消化されているかどうかを確認します。
3) 制限酵素処理したDNA を精製し,滅菌した水に溶かし,-20℃で保存します。
2.トランスフェクション
1) トランスフェクション前に2×106 細胞を60mm 培養ディッシュに抗生物質を加えずに播きます。16-24 時間後,70-80%コンフルになった時点でトランスフェクションを開始します。
2) トランスフェクション試薬(Fugene® やLipofectamineTM Plus など)を用いてトランスフェクション後,次の日にトランスフェクション試薬が含まれる培地をアスピレートし,4mL の完全培地を加えます。
3) プラークの状態をチェックしながら7 日間インキュベートします。
4) トランスフェクション後10 日にプラークを確認し,培養ができていれば(50%以上)クルードウイルスライセートを回収します。
※ 15 日以上の培養は行わないでください。
3.クルードウイルスライセートの回収
1) アデノウイルスを含む細胞を5-10mL の滅菌したピペットで,プレートからはがし,回収します。細胞と培地を滅菌したチューブに移します。
2) 凍結融解を3 回繰り返し(37℃のウォーターバスとドライアイス- メタノールバス)ウイルスを細胞からリリースさせます。
3) 細胞溶解液を卓上遠心機で3000rpm,15 分間室温で遠心し,細胞残渣をペレット化します。
4) 得られたクルードライセート(ウイルスストック)を-80℃で保存します。
4.ウイルスの増幅
下記手順はT75 フラスコで行うことを推奨します。
1) インフェクション前に3-5x106 細胞を播きます。
2) 上記のクルードライセート50%を培地に加えます。
3) インフェクション中の24-48 時間は,顕微鏡で cytopathic effects(CPE)をチェックします。CPE がほとんど完了したら(ほとんどの細胞が円状でフラスコに完全に接着していない状態)培地をピペッティングして細胞を採取し,フラスコ表面から培地中に感染細胞を移動させます。
4) 感染細胞と培地を集め,1000 x g,5 分で遠心し,細胞をペレット化させます。
5) 上清を取り除き,細胞ペレットを培地または10 mM Tris, pH 8.0, 100 mM NaCl に再懸濁させます。
6) 凍結融解によって細胞懸濁液からウイルスをリリースさせます。
3000 x g,10 分間で遠心し,得られた上清をウイルスストックとして保存します。
*ウイルス上清は-80℃に保存できますし,すぐに精製・タイター測定にも使用できます。
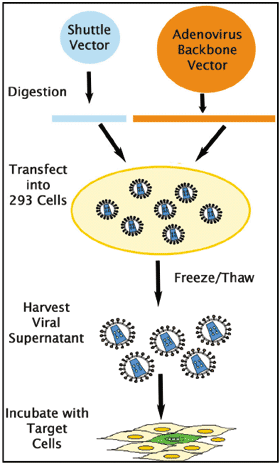
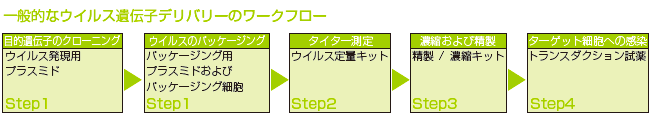
図1: アデノウイルス発現システムの概要
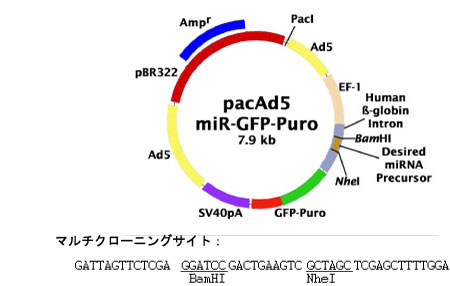
図2: pacAd ベクター

お問い合わせ
商品に関するご相談・お問い合わせ
![]()
受付時間:平日 (9:00〜17:30)
© COSMO BIO
