






















タンパク質はアミノ酸が重縮合したものであることから、各アミノ酸の含量すなわちアミノ酸組成はそのタンパク質に特有のパターンを示します。しかし、アミノ酸組成から得られる情報はかなり限定されたものです。たとえば食品タンパク質の場合の栄養価などです。その他には第2章で述べたように、構成アミノ酸残基の側鎖の性質から等電点や理論滴定曲線などのイオン的な性質が推定できます。これらはイオン交換クロマトグラフィーに用いる条件やネイティブ電気泳動での移動度の予測に用いられます。また、アミノ酸配列が知られているタンパク質のペプチドマッピングを行う場合に、逆相クロマトグラフィーでの溶出位置の予測に用いることも報告されています1)。一方で、タンパク質のアミノ酸配列から得られる情報は、アミノ酸組成だけから得られる情報に比べると、質・量ともに大きな差があり、以降に述べようにさまざまな解析ができます。
アミノ酸配列から得られるプロファイル
あるタンパク質について、アミノ酸配列が知られている場合、得られる情報は飛躍的に多くなります。たとえば、そのアミノ酸配列から疎水性部位を予測してそのプロフィールを作成することにより、目的配列がタンパク質の内側あるいは外側に存在するかを推定できます。特に膜タンパク質においては膜貫通部位の予測などに利用されます。親水性度(hydrophili¬ci¬ty)あるいはその逆の意味の疎水性度(hydro¬pho¬bicity)は、各アミノ酸について水と非極性溶媒との間の分配係数から求めた熱力学的な数値で、疎水性のより大きな基は負の疎水性度で示されます。しかし、側鎖とポリペプチド主鎖あるいは溶媒との相互作用などの影響が複雑に絡み合うので、当り障りの無いハイドロパシー(hydropathy)という言葉がよく使われます2)。なお、極性と非極性の両方の性質を示すセグメントを持つ場合があり、このような性質を両親媒性(amphi-philic)といいます。前章で紹介したアラインメントエディタを用いることによって、ここで述べた各種のプロフィールや次節で述べる抗原部位などについての情報を、アミノ酸配列から得ることができます。
2種のタンパク質のアミノ酸配列の類似性(ホモロジー)を調べる方法の一つに、Diagonal dot-plot3) (Harr Plot)があります。縦軸と横軸に、比較するそれぞれのタンパク質のアミノ酸配列を当てはめ、それぞれのアミノ酸残基が一致した交点にマークを記入したものです。着目するアミノ酸残基の前後でどの程度配列が一致しているかが直感的に分かる方法です。全く同じアミノ酸配列のタンパク質を縦軸と横軸に用いたこの方法でプロットした場合には、対角線が引かれます。
抗原となり得る部位の予測
第3章でも述べましたが、あるタンパク質に対する抗体を作製するためにペプチドを合成して抗原として用いなければならない場合があります。このような場合に、対象タンパク質について抗原となり得る部位を予測することが必要となり、その当たり外れが抗体産生の成否を決めることとなります。そのための目安(antigenicityあるいはantigenic index)として疎水性度の他に柔軟性4) (flexibility)、極性(polarity)、その部位の表面への露出のし易さ(surface probability)、さらには二次構造などの諸要素が用いられて総合的に判断することとなります。
タンパク質崩壊との関連
真核生物タンパク質のアミノ酸配列と、その細胞内での崩壊の半減期との関連が提唱されています。半減期が2時間以上のタンパク質には「PEST領域」5)すなわちPro, Glu, Ser, Thrを含む領域がほとんど見当たらないという報告や、N末端に位置するアミノ酸がin vivoでの半減期を決めるという「N-end rule」6)も提唱されていました。細胞内のプロテアソーム・ユビキチン系において、対象タンパク質にユビキチンを結合させる酵素がこれらの配列を認識すると考えられています。
細胞外に分泌されるタンパク質の場合は情況が異なります。血流中でラクトフェリンについては、ミルク由来のものと好中球由来のものでは糖鎖の組成が違うため(フコースの有無)、肝臓で選別されているという報告があります7)。
モチーフとドメイン
ある一定の機能を持つと期待される特徴的な共通の配列や構造をモチーフ(motif)といいます。モチーフ配列は3〜10個のアミノ酸残基から構成され、タンパク質の非構造領域に存在して明確な高次構造はとらないため8) 、正確にモチーフを予測することは困難と考えられています。しかし、タンパク質の生合成とその後の機能の調節にモチーフ配列が大きく関わっているために、モチーフ配列のより精度の高い予測法の研究が行われています。これまでに知られているモチーフには、遺伝子調節タンパク質のDNA結合モチーフであるホメオドメイン、ロイシンジッパーモチーフ、ジンクフィンガーモチーフ、ヘリックス-ループ-ヘリックス(HLH)モチーフ、ヘアピンβシートモチーフなどがあります。一方、ドメインは30残基以上で構成され、進化的に高度に保存された高次構造をとる配列です。ドメインとドメインとの相互作用は比較的強固ですが、ドメインとモチーフとの相互作用は弱いものが多いと考えられています8)。
高次構造の予測
ポリペプチド主鎖の構造、すなわち骨格構造は二次構造(secondary structure)と呼ばれ、規則的な繰り返し部分とそうでない部分とに分けられます。規則構造はαヘリックス、平行あるいは逆平行のβ構造(β-pleated sheet、β-structure)、βターンです。一般に球状のタンパク質分子表面の近くでは、ポリペプチド鎖の方向を変える折れ曲がり部分があり、ループ(loopまたはreverse turn、β-bend)などと呼ばれています。その中でβ-ヘアピン(β-hairpin)がよく知られており、逆平行β構造内で隣接するβストランドを繋いでいて、1残基から形成されている場合はγターン(g -turn)といわれます。2残基で形成されているβターン(β-turn)が一般的です。
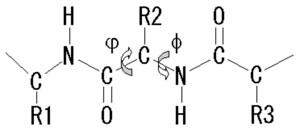
図12-1.ポリペプチド主鎖の回転角
アミノ酸残基間のペプチド結合、すなわちアミド結合(-NH-CO-)は平面構造をとっており、C-N結合の周りの回転はできません。その他の2種の結合の周りの回転は可能ですが、全く自由ではなく立体障害などである範囲の値しかとれません。なお、Cα-C結合の回転角はΨ(プサイ)、Cα-N結合の回転角はΦ(ファイ)です。これらの結合角の回転の制限とそれらの間の水素結合による安定化が主な要素となって二次構造が決まります。そこでペプチド鎖全体についてこれらの可能な角度を計算し、かつアミノ酸側鎖間の相互作用(水素結合、疎水結合)などを考慮して、エネルギー的に最も安定な構造を求めることから最終的に立体構造を推定出来ると考えられます。タンパク質のような巨大分子では現実的ではないと言われていましたが、最近は人工頭脳(AI)による立体構造の推定が現実味を帯びてきました(14章の補足を参照)。最も簡単な二次構造の推定方法はChou & Fasman法9)として知られている経験的な方法で、各アミノ酸について規則構造の形成し易さの指数から、その二次構造を推定するものです。たとえば、αヘリックスを形成し易いアミノ酸残基はGlu, Ala, Leu, Met, Gln, Lys, Arg, His、β構造(βストランド)を形成し易いものはVal, Ile, Tyr, Cys, Trp, Phe, Thr、ターンを形成し易いものはGly, Asn, Pro, Ser, Aspとなっています。さらにαヘリックス軸を紙面に垂直に投影した図を作ることによって、ヘリックスを構成しているアミノ酸残基の側鎖間の相互作用を推定することができます。これをhelical wheel(車輪投影モデル)3)といいます。
近年、膨大な数のゲノム解析データとタンパク質の配列データ、加えてタンパク質の立体構造データが多く蓄積され、さらこれらのデータを扱う高度なプログラムが開発されて、高精度の二次構造の予測が可能になり、現在では十分に実用的な方法になったと考えられています10)。詳細は他の成書や解説を参照して下さい。

