






















すでに述べましたが、タンパク質のアミノ酸配列を一次構造(primary structure)といい、各ペプチド主鎖がどのような構造をとっているかを二次構造(secondary structure)といいます。すなわち、αヘリックス、β構造、βターンのように規則性(繰り返し)のある構造と不規則構造です。これらがドメインと呼ばれるペプチド鎖の折りたたみ構造を形成し、タンパク質の分子全体の構造をとります。この時、ドメインよりさらに大きなローブ(lobe)構造をとって分子構造を形成する場合もあります。これを三次構造といいます。さらに、タンパク質分子が複数の会合体を形成して機能を発揮する場合があり、それを四次構造あるいはサブユニット構造といいます。これらタンパク質の高次構造は、その一次構造によって規定されていると信じて差し支え無いでしょう。
いわゆる未変性(native)の状態にあるタンパク質から、SDSや尿素などの変性剤、あるいはpHや温度条件によってその状態が変化することを変性(denaturation)といいます。ある場合にはこの変化は可逆的であり、そのような時にもとの状態にもどることを再生あるいは巻き戻し(renaturationあるいはrefolding)といいます。変性のメカニズムを知ることによって、逆にタンパク質の折りたたみに関する情報が得られる可能性があります。また、フォールディング(折りたたみ、folding)とは細胞内でタンパク質が生合成された場合に、ペプチド主鎖を折りたたんで構造をとることをいいます。タンパク質が立体構造をとる時、まず最初に二次構造の骨組みが出来てからその後に三次構造が形成されるというフレームワークモデルが提案され、この中間の状態をモルテングロビュール(molten globule)構造とよんでいます。変性の時にはこの逆の過程、すなわち未変性状態から中間状態を経て変性状態になると考えられています。
タンパク質の二次構造を推定するには、過去には旋光分散(ORD)測定が用いられていましたが、その後はもっぱら円偏光二色性(CD)スペクトル測定が用いられています。その他に赤外線スペクトル測定も有効です。また、タンパク質の三次構造の決定にはX線結晶構造解析が用いられ、ペプチドや比較的分子量の小さいタンパク質の場合には核磁気共鳴(NMR)による解析が有効です。
CDスペクトルの測定によって二次構造を推定することができるのは、αヘリックス、β構造、不規則構造のそれぞれで、紫外部領域でのスペクトルパターンとCD強度が異なるからです。合成ポリアミノ酸であるポリ-L-グルタミン酸あるいはポリ-L-リジンなどでは、pHを変えたり、加熱することにより二次構造が変わります。それらのCDスペクトルでは、αヘリックスの場合に208-209, 222 nmに負の極大、191-193 nmに正の極大、β構造では216-218 nmに負の極大、195-200 nmに正の極大、不規則構造では195-200 nmに負の極大が観察されています2)。
モデルペプチドではなく、実際に高次構造が知られている15種のタンパク質からαヘリックス、β構造、βターン、不規則構造にそれぞれ固有のCDスペクトルを求め(図13‐1)、その値を用いて対象タンパク質のCDスペクトルに最もよく適合する構造の組み合せを求める方法もあります。この方法を用いてウシラクトフェリンの紫外部でのCDスペクトル(図13-2)から二次構造のそれぞれの含量を計算しました。その結果、ウシラクトフェリンでは、αヘリックス 12%、β構造 37%、βターン 16%、不規則構造 34%と算定されました3)。
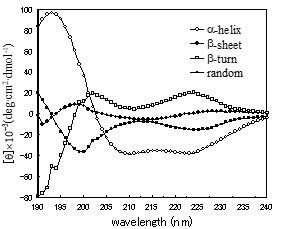
図13-1.高次構造の明らかな15種類のタンパク質から導かれた二次構造のCDスペクトル1)。
紫外部のCDスペクトル測定によって二次構造を推定する方法は、高分解能NMRやX線結晶構造解析に比べて少ない労力で測定・解析が出来るうえ、比較的に測定操作も簡便です。そのため、詳細な構造データが得られていないタンパク質の構造予測に関する情報を提供するうえで有効な方法です。なお、タンパク質は揺らいだ動的な構造をもつことが知られていますが、ほとんどの場合、溶液中と結晶での構造は同じと考えられています。また、赤外線領域(波数1600- 1700 cm-1)でのCDスペクトルは二次構造によるパターンがそれぞれ著しく異なるために、より詳しい情報が得られ、非常に有効と考えられています。
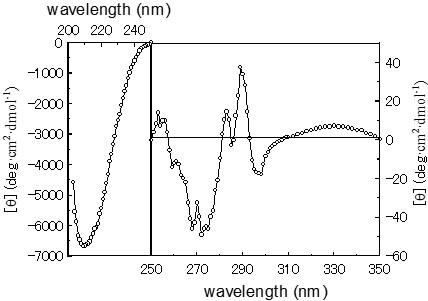
図13-2.ウシラクトフェリンのCDスペクトルの一例。左右で縦軸のスケールが違うことに注意。溶媒は0.05 M リン酸緩衝液 (pH 6.2)。日本分光(JASCO)model J-500で測定。)
250-300 nmの波長範囲で測定されるCDスペクトル(図12-2の右側)は、アミノ酸残基の芳香族側鎖に起因するもので、ポリペプチド主鎖の構造変化が生じていない場合でもより鋭敏に変化を検出できます。たとえば温度やpHの変化、変性剤添加、リガンドの結合などによって生ずる芳香族基の周辺の微細な環境変化が観察できます。これらは二次構造の変化を伴なわない微妙な変化に対応して検出されるため、ストップトフロー(stopped-flow)法やpHジャンプ(pH-jump)法を用いたタンパク質の構造形成の中間状態の研究、組換えタンパク質がもともとのタンパク質とどのように異なるかの比較などにも活用されています。また、発色団すなわち芳香族基による誘起CDスペクトルの測定をした例として、ブルー色素(Cibacron Blue F3GA)とラクトフェリンの相互作用を可視領域での誘起CDスペクトルから測定し、ウシとヒトとで色素との結合様式に違いがあることを検出できました4)(機能編、リガンドとの相互作用の章を参照)。
タンパク質の赤外線吸収スペクトルでは、波数(波長の逆数で表し、単位はcm-1)が1500〜1700の範囲にペプチド結合に特異的な吸収帯があり、特にアミド I (1600-1700 cm-1)、アミド II (1500-1550 cm-1)と呼ばれる振動帯が二次構造の推定に用いられています。これはアミノ酸残基が分子鎖に組み込まれた構造をとることで、各原子間の運動(伸縮、振動、回転)が変化し、吸収帯のシフトとして観測されるためです。αヘリックスでは1650 cm-1 と1546 cm-1 に、平行β構造では1630 cm-1 と1530 cm-1 に、逆平行β構造では1632 cm-1 と1530 cm-1 に、不規則コイルでは1655 cm-1 と1535 cm-1 に主な吸収が観察されます。すなわち吸収の生じた波長(波数)から分子の運動(振動数)の解析ができる訳です。
これらの波長領域では水の吸収が大きいため、溶媒には重水(D2O)を用います。一方、CDスペクトル測定とは違って、粉末やフィルム状の試料も測定できます。なお、赤外線吸収スペクトルでは時々刻々と変わる分子構造をピコ秒刻みで追跡することも可能です。
タンパク質溶液に振動数ν0のレーザー光を当て散乱光を測定した場合に、ν0 ± νi に観察される弱い散乱光をラマン散乱といいます。このようなタンパク質のラマン分光では、アミド I 、II、III の振動数で二次構造が分かり、S-SやC-S伸縮振動数でCys架橋やMet側鎖の構造、TyrやTrpのラマン線からそれらの環境や水素結合の有無を推定することが出来ます。また、励起させるレーザー光の波長を分子の吸収帯に近づけると、その吸収帯を与える発色団のラマン散乱が非常に強くなる現象があります。これを共鳴ラマン散乱といいます。
アミドII領域の赤外線吸収スペクトルやラマン分光あるいはNMR化学シフト法では、CDスペクトル測定よりも高濃度の試料が必要です。この場合、タンパク質同士の会合の影響がでることがあります。これらの要因のために、分光学的な方法によって推定される二次構造含量の精度には限界があります。
X線結晶構造解析では、タンパク質分子が規則正しく並んだ結晶(単結晶)にX線を照射して得られる回析X線の測定強度から、フーリエ変換で結晶中の電子密度分布を求め、さらに重原子(Hg, Ptなど)をタンパク質結晶内に導入する方法で位相を決め、最終的にタンパク質構造のモデルを構築します。タンパク質の質の良い単結晶が十分量確保できるかどうかがその成否を左右します。生体に含まれている量が少なく、ごく微量しか分離できなかったタンパク質でも、微生物などに組換えタンパク質を生産させて十分な量を得ることができるようになり、非常に多くのデータが蓄積してきました。
核磁気共鳴(NMR)は水溶液中のタンパク質の構造を推定するため、結晶化できないタンパク質の解析にも有効です。また、タンパク質の動的な構造の解析、フレキシブルで不規則な構造の解析にも適用できます。強い磁場に置かれたタンパク質分子中の水素原子(プロトン、1H)の核スピンの共鳴を観察する方法です。化学シフト、スピン結合、核オーバーハウザー効果などを測定する方法が開発され、立体構造の解析に用いられています。超高磁場磁石(プロトンの共鳴周波数で800 Hz)でも分子量が3万程度までのタンパク質しか解析できませんでしたが、最近ではさらに大きなタンパク質が扱えるようになってきています。アミノ酸が60個程度までのタンパク質では二次元DQFCOSY, TOCSY, NOESYの3種のスペクトルを測定し、より大きいタンパク質では15Nと13Cとで二重に標識して異種多次元NMR法で測定して主鎖の帰属を行っています。

