





















 このページを印刷する
このページを印刷する
チューブリンの過剰グルタミル化、ミトコンドリア、神経変性
微小管(MTs)は、哺乳類細胞の細胞骨格を構成する3つの必須タンパク質の1つで、α/β-チューブリンヘテロ二量体で構成されています。また、細胞の発生、増殖、運動性、機械的シグナル伝達、および細胞内輸送において非常に重要な役割を担っています。MTsの機能制御は少なくとも7種の翻訳後修飾(PTMs)によって行われます。翻訳後修飾は、通常は重合後に(動的に対して)安定なMTsのα/β-チューブリンヘテロ二量体で優先的に行われます。PTMsは高度に動的で、標的タンパク質のアミノ酸残基に化学基またはペプチドを付加することで、タンパク質の機能、結合パートナー、または細胞内局在化を制御する可逆的な過程でもあります1-4)。
様々な鎖長のグルタミン酸側鎖をグルタミン酸残基の初期配列に付加するポリグルタミル化PTMは、1990年代初頭に初めて報告されました。α-,β-チューブリンのC末端尾部(CTTs)のグルタミン酸残基が最も一般的な基質です1-6)(図1)。グルタミル化酵素はタンパク質のチューブリンチロシンリガーゼ様(TTLL)ファミリーのメンバーです1-4, 7, 8)。哺乳類細胞において、細胞質カルボキシペプチダーゼ(CCP;またはNna)はCCP1、4、5、および6と共に脱グルタミル化酵素として機能し、グルタミン酸側鎖を除去します。3つの酵素(CCP1、CCP4、およびCCP6)は、ポリグルタミン酸鎖の短縮反応を触媒するのに対し、CCP5は枝分かれ部位のグルタミン酸残基を特異的に除去します1-4, 9-11)(図1)。生理的なポリグルタミル化は、神経細胞の細胞体や神経突起(樹状突起や軸索)内のMTsを修飾し、MTを基盤とした様々な神経細胞機能を制御しています1-4)。
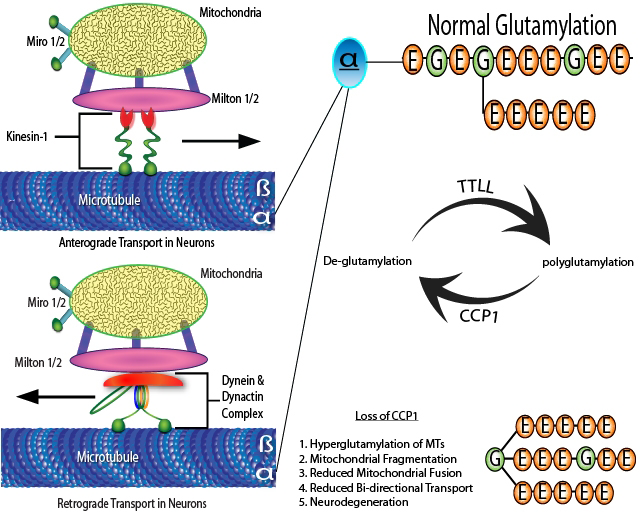
図1:双方向的輸送とCCP1活性欠損による過剰グルタミル化の効果
MTsに沿った輸送を駆動するために、順行性モーターキネシン-1と逆行性モーターダイニン/ダイナクチン複合体がミトコンドリア上でMiltonやMiroと直接相互作用する。脱グルタミル化酵素であるCCP1の機能欠損は、ミトコンドリア動態に負の影響を及ぼす過剰グルタミル化を引き起こし、さらに神経変性を引き起こす。チューブリンのカルボキシル末端のアミノ酸はTTLLs(グルタミル化酵素)やCCP1を始めとするCCPs(脱グルタミル化酵素)に作用を受けることがある。CCP1はポリグルタミン酸側鎖から枝分かれ部位までのグルタミン酸残基を切断する。CCP1の不活性化変異はpcdマウスにおけるプルキンエニューロン変性の原因となる。本図は参考文献9と17を改変した。
神経変性とミトコンドリア
チューブリンの過剰グルタミル化は神経変性の原因となることがあり、pcd(プルキンエ細胞変性)マウスにおいてCCP1の不活性化変異によるプルキンエ細胞死が観察されました9-12)。若いpcdマウス小脳では、チューブリン特異的神経細胞ポリグルタミル化酵素(TTLL1)の下方制御によりプルキンエ細胞死が防がれ、運動協調性が改善されました9)。プルキンエ細胞特異的Ccp1ノックアウトマウスを用いた近年の研究から、CCP1不活性化によるチューブリンの過剰グルタミル化が神経変性を引き起こすだけでなく、MTを基盤とした神経細胞内輸送の破壊も神経変性を引き起こしていることが確認されました13, 14)。Ccp1とttll1の両方を欠損するマウスではプルキンエ細胞死が起こりません13, 14)。さらに興味深いものに、pcdマウスにおいてCCP1発現/活性の欠損がどのように神経変性を引き起こすのかを明らかにした機構研究があります14, 15)(図1)。pcdマウスの初代培養小脳神経細胞とCCP1欠損網膜色素上皮細胞(pcdマウスの網膜変性に関与)では、予想通りチューブリンポリグルタミル化が増加しました14, 15)。さらに、Ccp1の欠損は、ミトコンドリア融合や双方向性(たとえば、順行性と逆行性)の運動性を減少させ、ミトコンドリアの断片化を増加させました14, 15)。他の神経変性の臨床前モデルと同様に、ヒト被験者における遺伝学的変異に関する知見は、神経変性疾患についてより深く学ぶだけでなく、治療的介入の考案という視点からも疾患研究の重要性を強めるものです。近年、Shashiら16) は、CCP1酵素活性の欠損(機能欠損型バリアントまたは単一アミノ酸置換によるミスセンスバリアントによる)がこれまで説明されていなかった小児期発症の進行性神経変性の原因であることを示しました。
以上をまとめると、上記の研究は、ミトコンドリアの神経細胞内輸送とミトコンドリアの機能、および形態維持がチューブリンの過剰グルタミル化により損なわれることを示しています。細胞内での双方向性輸送とイオン濃度勾配維持は、シナプス性神経伝達やシナプス小胞リサイクリングなどの基本的な神経機能に必要ですが、重要なことは、ミトコンドリアがこれらに必要なエネルギーを供給しているということです。ミトコンドリアは、その形態と数だけでなく、ミトコンドリアの機能と位置を制御するプロセスである、連続的な融合、核分裂、および輸送を受ける非常に動的なオルガネラです。ミトコンドリア自身は、特定の細胞内部位へ局在化する際には、MTsに沿った迅速な双方向性細胞内輸送に依存しています17)(図1)。モーターアダプター複合体は、主に順行性 モーターキネシン-1(または、キネシン重鎖 [KHC] または KIF5)、逆行性モーターダイニン(ダイナクチンとの複合体で)、Miro1と2、およびMilton1と2です17)。Miltonのアイソフォームは、はMT介在性輸送のために分子モーターをミトコンドリアに運搬するミトコンドリアアダプタータンパク質であり、Miroのアイソフォームは、はミトコンドリアRho GTPasesです。順行性 モーターキネシン-1と逆行性モーターダイニン/ダイナクチン複合体は、ミトコンドリア上でMiltonsやMirosと直接相互作用し、MTsに沿った双方向性移動を駆動します18, 19)。
ところで、これらの知見を他の神経変性疾患に拡張できるのか、という重要な疑問があります。MAPs(微小管関連タンパク質)の結合に影響するものをはじめとして、病的なチューブリンPTMsはパーキンソン病(PD)や様々なタウオパチーにおいて主要な役割を担うことが既によく知られています12, 20)。他の神経変性疾患におけるMTポリグルタミル化の役割は未だよくわかっていないものの、ミトコンドリア機能障害はPD、アルツハイマー病(AD)、ハンチントン病(HD)、および筋萎縮性側索硬化症(ALS)においても認められています17)。ミトコンドリアの融合や分裂に重要な制御因子(例えば、Drp1、OPA1、マイトフュージンなど)が同定され、神経変性疾患におけるミトコンドリア動態の役割を理解することの重要性がより強まっています。さらに、チューブリンPTMs、ミトコンドリア機能障害、および神経変性疾患のの間にある潜在的な相互作用を検討する必要性があることは明らかです。
まとめ
近年、チューブリンのポリ修飾に関する理解が進んでいるにも関わらず、グルタミル化/脱グルタミル化の非酵素的制御因子(例えば、CSAP [cilia and spindle-associated protein])21) やチューブリン脱グルタミラーゼの網羅的な同定、グルタミル化(および他のPTMs)がMAPs(例えば、タウ、分子モーターなど)への結合やMT切断酵素活性21) にどのような影響を及ぼすのかを完全に理解するためには、多くの研究課題が残っています9-11, 21)。グルタミル化酵素、脱グルタミル化酵素と関連する制御因子を全て同定することで、病態生理学的な特徴として細胞内輸送の機能障害を有する神経変性疾患に新たな治療標的を見出すことができるでしょう。研究者の方々をお手伝いするため、Cytoskeleton社では、内在性PTMsレベルが測定できるSignal-Seeker Detection Kits、チューブリン活性キットや結合アッセイキット、微小管用の生細胞イメージング用プローブ、および精製チューブリンタンパク質などをご提供しています。
参考文献
- Hammond J. et al. 2008. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 20, 71-76.
- Janke C. and Kneussel M. 2010. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 33, 362-372.
- Wloga D. and Gaertig J. 2010. Post-translational modifications of microtubules. J. Cell Sci. 123, 3447-3455.
- Wloga D. et al. 2017. Tubulin post-translational modifications and microtubule dynamics. Int. J. Mol. Sci. 18, 2207.
- Edde B. et al. 1990. Posttranslational glutamylation of alpha-tubulin. Science. 247, 83-85.
- Nogales E. 2000. Structural insights into microtubule function. Annu. Rev. Biochem. 69, 277-302.
- Janke C. et al. 2005. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 308, 1758-1762.
- van Dijk J. et al. 2007. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol. Cell. 26, 437-448.
- Rogowski K. et al. 2010. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 143, 564-578.
- Kimura Y. et al. 2010. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J. Biol. Chem. 285, 22936-22941.
- O’Hagan R. et al. 2011. The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr. Biol. 21, 1685-1694.
- Baird F.J. and Bennett C.L. 2013. Microtubule defects and neurodegeneration. J. Genet. Syndr. Gene Ther. 4, 11.
- Magiera M.M. et al. 2018. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 37, e100440.
- Strzyz P. 2018. Neurodegenerative polyglutamylation. Nat. Rev. Mol. Cell Biol. 20, 1.
- Gilmore-Hall S. et al. 2018. CCP1 promotes mitochondrial fusion and motility to prevent Purkinje cell neuron loss in pcd mice. J. Cell Biol. 218, 206-219.
- Shashi V. et al. 2018. Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J. 37, e100540.
- Gao J. et al. 2017. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. 6, 25.
- Guo X. et al. 2005. The GTPase dmiro is required for axonal transport of mitochondria to drosophila synapses. Neuron. 47, 379?393.
- Glater E.E. et al. 2006. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545?557.
- Pellegrini L. et al. 2017. Back to the tubule: microtubule dynamics in Parkinson’s disease. Cell. Mol. Life Sci. 74, 409-434.
- van der Laan S. et al. 2019. Tubulin glutamylation: a skeleton key for neurodegenerative diseases. Neural Regen. Res. 14, 1899-1900.
蛍光標識チューブリンタンパク質
[商品詳細]
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 |
|---|---|---|---|---|
Tubulin, Porcine, 7-amino-4-methylcoumarin-3-acetic acid |
CYT | TL440M-A | 5*20 UG |
¥105,000 |
| Tubulin, Porcine, 7-amino-4-methylcoumarin-3-acetic acid |
CYT | TL440M-B | 20*20 UG |
¥266,000 |
| Tubulin, Porcine, HiLyte FluorTM 488 |
CYT | TL488M-A | 5*20 UG |
¥105,000 |
| Tubulin, Porcine, HiLyte FluorTM 488 |
CYT | TL488M-B | 20*20 UG |
¥266,000 |
| Tubulin, Porcine, Tetramethylrhodamine |
CYT | TL590M-A | 5*20 UG |
¥105,000 |
| Tubulin, Porcine, Tetramethylrhodamine |
CYT | TL590M-B | 20*20 UG |
¥266,000 |
| Tubulin, Porcine, HiLyte FluorTM 647 |
CYT | TL670M-A | 5*20 UG |
¥105,000 |
| Tubulin, Porcine, HiLyte FluorTM 647 |
CYT | TL670M-B | 20*20 UG |
¥266,000 |

Signal Seeker™ キット
[商品詳細]
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 |
|---|---|---|---|---|
| Signal-SeekerTM Phosphotyrosine Detection Kit | CYT | BK160 | 30 ASSAY |
¥221,000 |
| Signal-SeekerTM Phosphotyrosine Detection Kit | CYT | BK160-S | 10 ASSAY |
¥112,000 |
| Signal-SeekerTM Ubiquitin Detection Kit | CYT | BK161 | 30 ASSAY |
¥221,000 |
| Signal-SeekerTM Ubiquitin Detection Kit | CYT | BK161-S | 10 ASSAY |
¥112,000 |
| Signal-SeekerTM SUMOylation Detection Kit | CYT | BK162 | 30 ASSAY |
¥221,000 |
| Signal-SeekerTM SUMOylation Detection Kit | CYT | BK162-S | 10 ASSAY |
¥112,000 |
| Signal-SeekerTM SUMOylation-1 Detection Kit | CYT | BK165 | 1 KIT [30 assays] |
¥221,000 |
| Signal-SeekerTM SUMOylation-1 Detection Kit | CYT | BK165-S | 1 KIT [10 assays] |
¥112,000 |
| Signal-SeekerTM Acetyl-Lysine Detection Kit |
CYT | BK163 | 30 ASSAY |
¥210,000 |
| Signal-SeekerTM Acetyl-Lysine Detection Kit |
CYT | BK163-S | 10 ASSAY |
¥112,000 |
チューブリンキット
[商品詳細]
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 |
|---|---|---|---|---|
| Tubulin Polymerization Assay Kit, Porcine |
CYT | BK004P | 1 KIT [24 assays] |
¥193,000 |
| Tubulin Polymerization Assay Kit |
CYT | BK006P | 1 KIT [24 assays] |
¥274,000 |
| Tubulin Polymerization Assay |
CYT | BK011P | 1 KIT [96 assays] |
¥280,000 |
| Microtubule Binding Protein Spin Down Assay Kit |
CYT | BK029 | 1 KIT [30-100 assays] |
¥231,000 |
Live Cell Imaging Products
[商品詳細]
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 |
|---|---|---|---|---|
| SiR-Tubulin Kit |
CYT | CY-SC002 | 1 KIT [50-300 slides] |
¥170,000 |
| SiR700-Tubulin Kit |
CYT | CY-SC014 | 1 KIT [35-200 slides] |
¥170,000 |
■ CYTOSKELETON NEWS バックナンバー
- 2020年10月号 紡錘体 - 可視化に向けた新規ツール
- 2020年8月号 細胞膜染色用蛍光プローブ
- 2020年4月号 生細胞におけるF-アクチンプローブ
- 2020年3月号 コロナウイルスと細胞骨格
- 2020年2月号 タウ(Tau)の将来性をMapping
- 2020年1月号 Rho GTPaseによる細胞遊走制御
- 2019年12月号 表現型プロファイリング:アクチンに焦点を当てたがん治療
- 2019年11月号 チューブリンの過剰グルタミル化、ミトコンドリア、神経変性
- 2019年9月号 細胞運動性を制御するために相互作用する膜張力とアクチン細胞骨格
- 2019年8月号 Rac1B、がん、およびRac1
- 2019年7月号 Rhoファミリー GTPases、神経可塑性、およびうつ状態
- 2019年6月号 アクチンメチオニン酸化: 動的制御の次の段階
- 2019年5月号 ミクログリアと神経変性疾患
- 2019年2月号 生細胞画像化に対するCNS疾患や障害
2018年
- 2018年12月号 アクチン細胞骨格とメカノトランスダクション(機械的シグナル伝達)
- 2018年11月号 軸索再生と細胞骨格
- 2018年10月号 ニューロンにおける微小管と極性
- 2018年8月号 Rab GTPase と 神経変性
- 2018年7月号 SUMO レスリング: バランスが全て
- 2018年6月号 なぜ K-Ras は発がん特異性を示すのか?
- 2018年5月号 治療標的としてのユビキチンプロテアソームシステム:チューブリンは関与するか?
- 2018年4月号 RhoファミリーGEFと樹状突起スパインの構造的可塑性
- 2018年3月号 βカテニンとTFC/LEF-1の翻訳後修飾による標準的なWntシグナル制御
- 2018年2月号 がん抑制遺伝子p53の翻訳後修飾による機能の調整
- 2018年1月号 自閉スペクトラム症におけるGEF Trioの役割
2017年
- 2017年12月号 プロフィリン: アクチン結合タンパク質の多機能な役割
- 2017年11月号 ミトコンドリアにおけるアセチル化:新たな考え方と治療への応用の可能性
- 2017年9月号 翻訳後修飾のアセチル化による微小管の安定化
- 2017年8月号 神経軸索におけるアクチンリングを基盤とした周期的膜骨格(PMS)
- 2017年7月号 E3ユビキチンリガーゼMdm2によるがん抑制遺伝子p53の翻訳後制御
- 2017年6月号 多能性幹細胞(PSC)での転写因子による翻訳後制御
- 2017年5月号 Arf6 GEF と癌細胞の浸潤・転移
- 2017年4月号 PTEN(Phosphatase and Tensin Homolog)による翻訳後制御
- 2017年3月号 Tau の翻訳後修飾: アルツハイマー病の治療標的
- 2017年2月号 樹状細胞の移動におけるアクチン結合タンパク質とF-アクチン
2016年
- 2016年11月/12月号 GEF を介した GTPase シグナル伝達の低分子阻害剤
- 2016年9月号 FtsZ タンパク質: 抗菌薬の新規ターゲット
- 2016年7月号 翻訳後修飾(PTM)は心臓病において細胞骨格タンパク質を調節する
- 2016年6月号 モータータンパク質キネシンと神経変性
- 2016年5月号 チロシンリン酸化は Rhoファミリー GTPase 活性を調節する
- 2016年4月号 Rac1と糖尿病: ポジティブな役割とネガティブな役割
- 2016年3月号 SUMO化: 細胞骨格タンパク質の機能を調節するレギュレーター
- 2016年1月/2月号 ビメンチン中間径フィラメント: リン酸化による調節
2015年
- 2015年8月号 タンパク質調節に不可欠な翻訳後修飾
- 2015年7月号 アクチン細胞骨格のライブセルイメージング
- 2015年6月号 有糸分裂に関わるタンパク質のSUMO化: 局在と機能
- 2015年5月号 Ras 癌の治療: 5つの有望なターゲット
- 2015年4月号 Ras 依存性の癌で注目される YAP1
- 2015年3月号 増刊号 統合失調症において遺伝子変異により誘導されるアクチン依存のシナプスの変化
- 2015年3月号 Ral GTPase を調節する翻訳後修飾
- 2015年1月/2月号 RhoA のリン酸化はシグナル伝達を調節する
- 2015年1月号 増刊号 微小管を不安定化する suprafenacine: 新規抗癌剤のリード化合物としての可能性
2014年
- 2014年12月号 増刊号 RhoA は心筋細胞におけるアクチン細胞骨格の再構成とグルコース取り込みを仲介する
- 2014年11月号 増刊号 樹状突起の形態形成: ドーパミンD1受容体 および Rho ファミリー GTPase による制御
- 2014年11月/12月号 GTPase 活性化アッセイ: アイソフォームの検出
- 2014年10月号 アルギニンの正電荷を消失させるシトルリン化
- 2014年9月号 キネシンサブドメインの探索
- 2014年9月号 増刊号 アクチン結合タンパク質コフィリンの S-ニトロシル化: 細胞移動に対する影響
- 2014年8月号 増刊号 原発性硬化性胆管炎における N-Ras 発現および活性
- 2014年8月号 SUMO化: 細胞骨格タンパク質を標的とした翻訳後修飾
- 2014年7月号 Sos/K-Ras 結合を介して Ras シグナル伝達を制御する新しい低分子阻害剤
- 2014年6月号 増刊号 頭頸部扁平上皮癌における microRNA-138 による RhoC のダウンレギュレーション
- 2014年6月号 Rho GTPase と活性酸素種: クロストークとフィードバック
- 2014年5月号 ミオシンのアセチル化はサルコメアの構造と機能を調節する
- 2014年4月号 リジンのアセチル化 - 多様な細胞プロセスの制御因子
- 2014年3月号 インテグリンを介したβ-アクチンの酸化還元制御: PDIの出現
- 2014年1/2月号 ダイニン: 一つのモーターが関わる複数の神経変性疾患
2013年
- 2013年11/12月号 ダイニン:チームとして強力に作用するモータータンパク質
- 2013年10月号 神経変性:Rhes、SUMO化、ハンチントン病
- 2013年9月号 モノユビキチン化:タンパク質調節のダイナミックなタグ
- 2013年8月号 Ras及びRhoのプレニル化による翻訳後修飾:癌創薬における役割
- 2013年7月号 アクチンが引き起こす膜突起による浸潤:コルタクチン
- 2013年6月号 アクチン修飾と細胞骨格
- 2013年5月号 微小管内部の実体
- 2013年4月号 神経変性におけるTauの多面性
- 2013年3月号 蛍光フィブロネクチンタンパク質を用いた特発性肺線維症の創薬
- 2013年1/2月号 樹状突起棘:発生におけるArf6の役割
2012年
- 2012年11/12月号 ミオシンの小分子モジュレーター
- 2012年10月号 Rhoファミリーパスウェイのユビキチン化と制御
- 2012年9月号 神経変性におけるRac1 GTPaseの機能
- 2012年8月号 上皮間葉転換(EMT)とRhoファミリー低分子量G-タンパク質の関与
- 2012年7月号 チューブリンの多重修飾:グルタミル化とグリシル化
- 2012年6月号 細胞接着のフィブロネクチン制御と原線維形成
- 2012年5月号 アクチン酸化サイクルの機能
- 2012年4月号 トラフィッキング:ArfとCdc42/Racの結合
- 2012年3月号 G-LISAを用いた心臓研究: 糖尿病性心筋症におけるRho経路に関する研究
- 2012年1月/2月号 FtsZ: 新たな抗生剤の標的となるチュ−ブリンホモログ
商品は「研究用試薬」です。人や動物の医療用・臨床診断用・食品用としては使用しないように、十分ご注意ください。
※ 表示価格について








 中身を見る
中身を見る 中身を見る
中身を見る 中身を見る
中身を見る
 中身を見る
中身を見る