





















 このページを印刷する
このページを印刷する
記事ID : 35768
ミクログリアと神経変性疾患 CYTOSKELETON NEWS 2019年5月号
ミクログリアと神経変性疾患
ミクログリアは初代免疫系細胞であり、哺乳動物の脳において、いわゆる貪食を専門とする細胞とよばれています。ミクログリアは安静状態の脳全体を絶えず探査しており、神経毒性のある病原体や損傷した細胞機構を検出すると、それらを排除するために活性化されます1, 2(図1)。健常なニューロンでは、ミクログリアは、ニューロン新生、神経発生、および神経可塑性といった重要な細胞機能にも寄与します3, 4。これらと同様に重要なこととして、パーキンソン病(PD: レビー小体)とアルツハイマー病(AD: Aβ斑)といった、病原性の誤って折り畳まれたタンパク質の凝集で特徴付けられる神経変性疾患におけるミクログリアの役割が挙げられます。ミクログリア媒介性の神経炎症は、PDやADのヒト脳やin vivo動物モデル脳において一般的な病態生理です1, 2, 4-9。さらに、近年の遺伝学的かつトランスクリプトーム研究より、ミクログリア関連シグナル伝達カスケードがAD病態形成において非常に重要な役割を担うことが明らかになりました1, 7。本ニュースレターでは、PDやADにおけるミクログリアの活性化と神経炎症についてお伝えします。
PDは、黒質緻密部におけるドーパミン(DA)ニューロンの損失、これに伴う脳幹神経節全体におよぶドーパミン作動性(DAergic)終末の損失、および主に細胞内α-シヌクレイン凝集からなるレビー小体で特徴付けられます。レビー小体はPDの病態生理的な特質であり、それ自身もPDの神経病理的相関物であるミクログリア媒介性神経炎症を惹起します1, 2, 8, 10(図1)。PD疾患の脳では、レビー小体神経突起は活性化ミクログリアと関連し、α-シヌクレイン沈着物は炎症性マーカーと相関します。In vitro細胞培養やin vivo動物モデルにおいても、α-シヌクレイン凝集とミクログリア活性化の関係が実証されています2, 8, 10。In vitroでは、α-シヌクレイン濃度依存性でミクログリアを活性化し、ミクログリアは貪食作用を介してα-シヌクレイン凝集体を除去するものの、これら2つの過程の正確な関係性は未だ解っていません10。マウスにおいて、野生型または変異体α-シヌクレインを過剰発現させると、早期のミクログリア活性化が引き起こされます。野生型ヒトα-シヌクレインをマウスPDモデル内でin vivo過剰発現させると、早期ミクログリア活性化とその持続、および複数の炎症促進性サイトカイン(例:腫瘍壊死因子α [TNF-α]、IL-1b、IL-6、およびIL-18等のインターロイキン [IL])やケモカインの放出増大が見られます2, 6。α-シヌクレイン発現は脳全体を包含するものの、この炎症性応答は黒質線条体経路からなる中脳DAニューロンに限定されます2, 6, 8。PDのMPTPマウスモデルにおいて、MPTP毒素はミクログリア媒介性炎症を誘発し、MPTP誘導性ドーパミン作動性のニューロン神経変性が生ずる前に炎症促進性サイトカインの予想された増加が生じます8。
ADの一般的な病理組織学的特徴として、Aβ斑の細胞外沈着と細胞内高リン酸化タウ濃縮体が挙げられます。ミクログリア活性化と炎症促進性サイトカイン放出、および続いて起こる神経炎症もまた、ADの病理学的特徴です1, 2, 7(図1)。数々の研究より、ADにおけるミクログリアと関連タンパク質の神経保護と神経毒性の役割が確認できています。一方で、病原性Aβ可溶性種と不溶性斑は食作用を経て、AD後期段階ではミクログリアが斑を取り囲み圧縮します(図1)。圧縮により現存の斑上へのさらなる沈着を防げる可能性があります1, 2, 7。さらに、近年の遺伝学研究より複数のAD危険性遺伝子が明らかとなり、このうち多くのものは直接的または間接的にミクログリアに関連します(例えば、ミエロイド細胞に発現するトリガー受容体2 [TREM2] および補体受容体1 [CR1] 遺伝子)1, 7。TREM2は食作用性シグナル伝達やミクログリア媒介性神経保護に必要であり、その機能障害によりADの病態生理が悪化します1, 2, 7。健常なニューロンにおいて、CR1や自然免疫系の補体シグナル伝達カスケードの他のメンバーは、食作用、免疫複合体の除去、および補体システムの負の制御においてミクログリアの支援を行い11, 12、神経回路の発生、成熟、および精密化の間に余剰シナプスの除去(つまり、シナプス剪定)を補助します13-15。神経毒性は、ヒトAD脳やマウスADモデルにおいて補体タンパク質が上方制御され活性化されると生じます13-16。興味深いことに、Aβ斑沈着が生ずる前でミクログリア機能障害が見られる際に上方制御が生じます13-16。これらの知見より、機能障害性ミクログリアと補体タンパク質が生理的シナプス剪定過程の機能不全による早期シナプス損失に寄与することが示唆されます1, 2, 16(図1)。AD関連シナプス損失とシナプス機能障害は、補体シグナル伝達を阻害すると救出されます1, 16。シナプス損失はAD(および類似疾患)における早期病理現象であり、認知機能低下と非常に強く相関することから、これらは注目すべきデータといえます17, 18。個別のADマウスモデルによるin vivo二光子顕微鏡観察より、Aβ斑の数が増大するとミクログリア機能が低下することが示されているように、ミクログリア機能障害は継続的な斑形成により悪化することが示唆されています19。
ミクログリアとミクログリア関連タンパク質はタウ病理学に関連付けられています。in vivoマウスモデルでは、未知経路を介して、補体タンパク質の活性化がタウ病態を悪化させると考えられます20, 21。さらに、in vivoタウ遺伝子導入マウスモデルにおいて、ミクログリアの過剰活性化がタウ病理の発生と進行を悪化させることが示されています22, 23。タウ変異体病理のin vivoモデルより、毒性タウ種によるミクログリアの取込みとエキソソームによるタウ放出が、病原性タウの細胞間伝達に寄与することが示唆されています7, 24。
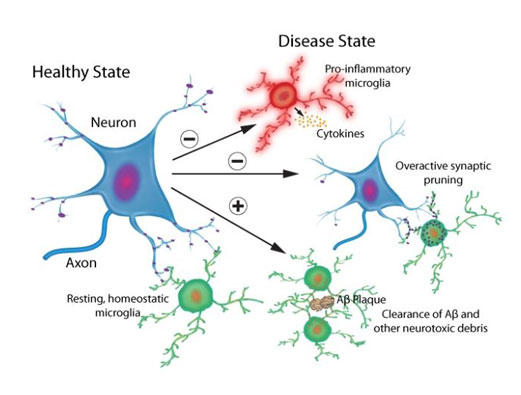
図1. 生理学的かつ病理学的ミクログリアプロセス
健常または病的なニューロンにおいて、ミクログリア媒介性シグナル伝達カスケードが神経保護性/神経栄養性および神経毒性効果を引き起こす。
まとめ
ミクログリアは脳において多様な生理的かつ病理的機能を示すため、多数の課題を提示しています。神経変性疾患では、いつ、どのようにして、抗炎症性から炎症促進性シグナル伝達への転換が生ずるのかはいまだに解明されていません。2つの対立するミクログリア表現型の変化の順序や頃合いを理解することは複雑なことであり、新しい研究ツールが必要となるでしょう25。実際、近年ではカルシウム指示薬を利用した生細胞画像が進歩しており、ニューロン機能やその損傷におけるミクログリアのin vivoにおける役割をよりよく理解する機会が得られています26。Cytoskeleton社 では、哺乳動物脳におけるミクログリア関連シグナル伝達カスケードを解読を目的とした、生細胞画像試薬、生成細胞骨格タンパク質、機能分析キット、および標的タンパク質の翻訳後修飾を定量するためのSignal-Seeker™ kitsをご提供しています。
参考文献
- Salter M.W. and Stevens B. 2017. Microglia emerge as central players in brain disease. Nat. Med. 23, 1018-1027
- Wolf S.A. et al. 2017. Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619-643.
- Bilimoria P.M. and Stevens B. 2015. Microglia function during brain development: New insights from animal models. Brain Res. 1617, 7-17.
- Sominsky L. et al. 2018. Microglia: Key players in neurodevelopment and neuronal plasticity. Int. J. Biochem. Cell Biol. 94, 56-60.
- Shen Z. et al. 2017. Microglia-targeted stem cell therapies for Alzheimer disease: a preclinical data review. J. Neurosci. Res. 95, 2420-2429.
- Lee S.-H. and Suk K. 2018. Kinase-based taming of brain microglia toward disease-modifying therapy. Front. Cell. Neurosci. 12, 474.
- Hansen D.V. et al. 2018. Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459-472.
- Subramaniam S. and Federoff J. 2017. Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front. Aging Neurosci. 9, 176.
- Ransohoff R.M. 2016. How neuroinflammation contributes to neurodegeneration. Science. 353, 777-783.
- Zhang Q.S. et al. 2017. Pathological alpha-synuclein exacerbates the progression of Parkinson’s disease through microglial activation. Toxicol. Lett. 265, 30-37.
- Khera R. and Das N. 2009. Complement Receptor 1: disease associations and therapeutic implications. Mol. Immunol. 46, 761-772.
- Fonseca M.I. et al. 2016. Analysis of the putative role of CR1 in Alzheimer’s disease: genetic association, expression, and function. PLoS ONE 11, e0149792.
- Schafer D.P. et al. 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 74, 691-705.
- Stephan A.H. et al. 2012. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369-389.
- Stevens B. et al. 2007. The classical complement cascade mediates CNS synapse elimination. Cell. 131, 1164-1178.
- Hong S. et al. 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 352, 712-716.
- Terry R.D. et al. 1991. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572-580.
- DeKosky S.T. et al. 1996. Structural correlates of cognition in dementia: quantification and assessment of synaptic change. Neurodegeneration. 5, 417-421.
- Krabbe G. et al. 2013. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PloS ONE. 8, e60921.
- Fonseca M.I. et al. 2009. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 183, 1375-1383.
- Britschgi M. et al. 2012. Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. J. Neuroinflammation. 9, 220.
- Bhaskar K. et al. 2010. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 68, 19-31.
- Maphis N. et al. 2015. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 138, 1738-1755.
- Asai H. et al. 2015. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 1584-1593.
- Tang Y. and Le W. 2015. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181-1194.
- Brawek B. et al. 2017. A new approach for ratiometric in vivo calcium imaging of microglia. Sci. Rep. 7, 6030.
Actin Products
[商品詳細]
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 |
|---|---|---|---|---|
Actin, Bovine, Unlabeled |
CYT | AD99-A | 1*1 MG |
¥44,000 |
| Actin, Bovine, Unlabeled |
CYT | AD99-B | 5*1 MG |
¥181,000 |
| Actin, Chicken |
CYT | AS99-A | 1*1 MG |
¥44,000 |
| Actin, Chicken |
CYT | AS99-B | 5*1 MG |
¥181,000 |
| Actin protein (pre-formed filaments), Rabbit |
CYT | AKF99-A | 1*1 MG |
¥69,000 |
| Actin protein (pre-formed filaments), Rabbit |
CYT | AKF99-B | 5*1 MG |
¥240,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL95-B | 1*1 MG |
¥35,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL95-C | 5*1 MG |
¥137,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL99-A | 4*250 UG |
¥73,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL99-B | 2*1 MG |
¥87,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL99-C | 5*1 MG |
¥177,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL99-D | 10*1 MG |
¥335,000 |
| Actin, Rabbit, Unlabeled |
CYT | AKL99-E | 20*1 MG |
お問い合わせ |
| Actin, Human, Unlabeled |
CYT | APHL99-A | 2*250 UG |
¥79,000 |
| Actin, Human, Unlabeled |
CYT | APHL99-C | 1*1 MG |
¥133,000 |
| Actin, Human, Unlabeled |
CYT | APHL99-E | 5*1 MG |
お問い合わせ |
| Actin, Human, Rhodamine Isothiocyanate |
CYT | APHR-A | 4*10 UG |
¥108,000 |
| Actin, Human, Rhodamine Isothiocyanate |
CYT | APHR-C | 20*10 UG |
¥339,000 |
| Actin, Rabbit, Rhodamine Isothiocyanate |
CYT | AR05-B | 10*20 UG |
¥104,000 |
| Actin, Rabbit, Rhodamine Isothiocyanate |
CYT | AR05-C | 20*20 UG |
¥200,000 |
| SiR-Actin Kit |
CYT | CY-SC001 | 1 KIT [50-300 slides] |
¥182,000 |
| SiR700-Actin Kit |
CYT | CY-SC013 | 1 KIT [35-200 slides] |
¥182,000 |
| Acti-stain 488 phalloidin, Plant |
CYT | PHDG1-A | 1*500 UL [300 slides] |
¥79,000 |
| Acti-stain 555 phalloidin, Mushroom |
CYT | PHDH1-A | 1*500 UL [300 slides] |
¥79,000 |
| Acti-stain 670 phalloidin, Plant |
CYT | PHDN1-A | 1*500 UL [300 slides] |
¥79,000 |
| Rhodamine Phalloidin, Rhodamine Isothiocyanate |
CYT | PHDR1 | 1*500 UL [300 slides] |
¥79,000 |
Actin Biochem Kits
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 |
|---|---|---|---|---|
| Actin Binding Protein Spin-Down Assay Biochem Kit, Rabbit |
CYT | BK001 | 1 KIT [30-100 assays] |
¥268,000 |
| Actin Binding Protein Spin-Down Assay Biochem Kit, Human |
CYT | BK013 | 1 KIT [30-100 assays] |
¥272,000 |
| Actin Polymerization Biochem Kit (fluorescence format), Rabbit |
CYT | BK003 | 1 KIT [30-100 assays] |
¥304,000 |
| G-Actin/F-actin In Vivo Assay Biochem Kit, Mouse |
CYT | BK037 | 1 KIT [30-100 assays] |
¥264,000 |
■ CYTOSKELETON NEWS バックナンバー
- 2020年10月号 紡錘体 - 可視化に向けた新規ツール
- 2020年8月号 細胞膜染色用蛍光プローブ
- 2020年4月号 生細胞におけるF-アクチンプローブ
- 2020年3月号 コロナウイルスと細胞骨格
- 2020年2月号 タウ(Tau)の将来性をMapping
- 2020年1月号 Rho GTPaseによる細胞遊走制御
- 2019年12月号 表現型プロファイリング:アクチンに焦点を当てたがん治療
- 2019年11月号 チューブリンの過剰グルタミル化、ミトコンドリア、神経変性
- 2019年9月号 細胞運動性を制御するために相互作用する膜張力とアクチン細胞骨格
- 2019年8月号 Rac1B、がん、およびRac1
- 2019年7月号 Rhoファミリー GTPases、神経可塑性、およびうつ状態
- 2019年6月号 アクチンメチオニン酸化: 動的制御の次の段階
- 2019年5月号 ミクログリアと神経変性疾患
- 2019年2月号 生細胞画像化に対するCNS疾患や障害
2018年
- 2018年12月号 アクチン細胞骨格とメカノトランスダクション(機械的シグナル伝達)
- 2018年11月号 軸索再生と細胞骨格
- 2018年10月号 ニューロンにおける微小管と極性
- 2018年8月号 Rab GTPase と 神経変性
- 2018年7月号 SUMO レスリング: バランスが全て
- 2018年6月号 なぜ K-Ras は発がん特異性を示すのか?
- 2018年5月号 治療標的としてのユビキチンプロテアソームシステム:チューブリンは関与するか?
- 2018年4月号 RhoファミリーGEFと樹状突起スパインの構造的可塑性
- 2018年3月号 βカテニンとTFC/LEF-1の翻訳後修飾による標準的なWntシグナル制御
- 2018年2月号 がん抑制遺伝子p53の翻訳後修飾による機能の調整
- 2018年1月号 自閉スペクトラム症におけるGEF Trioの役割
2017年
- 2017年12月号 プロフィリン: アクチン結合タンパク質の多機能な役割
- 2017年11月号 ミトコンドリアにおけるアセチル化:新たな考え方と治療への応用の可能性
- 2017年9月号 翻訳後修飾のアセチル化による微小管の安定化
- 2017年8月号 神経軸索におけるアクチンリングを基盤とした周期的膜骨格(PMS)
- 2017年7月号 E3ユビキチンリガーゼMdm2によるがん抑制遺伝子p53の翻訳後制御
- 2017年6月号 多能性幹細胞(PSC)での転写因子による翻訳後制御
- 2017年5月号 Arf6 GEF と癌細胞の浸潤・転移
- 2017年4月号 PTEN(Phosphatase and Tensin Homolog)による翻訳後制御
- 2017年3月号 Tau の翻訳後修飾: アルツハイマー病の治療標的
- 2017年2月号 樹状細胞の移動におけるアクチン結合タンパク質とF-アクチン
2016年
- 2016年11月/12月号 GEF を介した GTPase シグナル伝達の低分子阻害剤
- 2016年9月号 FtsZ タンパク質: 抗菌薬の新規ターゲット
- 2016年7月号 翻訳後修飾(PTM)は心臓病において細胞骨格タンパク質を調節する
- 2016年6月号 モータータンパク質キネシンと神経変性
- 2016年5月号 チロシンリン酸化は Rhoファミリー GTPase 活性を調節する
- 2016年4月号 Rac1と糖尿病: ポジティブな役割とネガティブな役割
- 2016年3月号 SUMO化: 細胞骨格タンパク質の機能を調節するレギュレーター
- 2016年1月/2月号 ビメンチン中間径フィラメント: リン酸化による調節
2015年
- 2015年8月号 タンパク質調節に不可欠な翻訳後修飾
- 2015年7月号 アクチン細胞骨格のライブセルイメージング
- 2015年6月号 有糸分裂に関わるタンパク質のSUMO化: 局在と機能
- 2015年5月号 Ras 癌の治療: 5つの有望なターゲット
- 2015年4月号 Ras 依存性の癌で注目される YAP1
- 2015年3月号 増刊号 統合失調症において遺伝子変異により誘導されるアクチン依存のシナプスの変化
- 2015年3月号 Ral GTPase を調節する翻訳後修飾
- 2015年1月/2月号 RhoA のリン酸化はシグナル伝達を調節する
- 2015年1月号 増刊号 微小管を不安定化する suprafenacine: 新規抗癌剤のリード化合物としての可能性
2014年
- 2014年12月号 増刊号 RhoA は心筋細胞におけるアクチン細胞骨格の再構成とグルコース取り込みを仲介する
- 2014年11月号 増刊号 樹状突起の形態形成: ドーパミンD1受容体 および Rho ファミリー GTPase による制御
- 2014年11月/12月号 GTPase 活性化アッセイ: アイソフォームの検出
- 2014年10月号 アルギニンの正電荷を消失させるシトルリン化
- 2014年9月号 キネシンサブドメインの探索
- 2014年9月号 増刊号 アクチン結合タンパク質コフィリンの S-ニトロシル化: 細胞移動に対する影響
- 2014年8月号 増刊号 原発性硬化性胆管炎における N-Ras 発現および活性
- 2014年8月号 SUMO化: 細胞骨格タンパク質を標的とした翻訳後修飾
- 2014年7月号 Sos/K-Ras 結合を介して Ras シグナル伝達を制御する新しい低分子阻害剤
- 2014年6月号 増刊号 頭頸部扁平上皮癌における microRNA-138 による RhoC のダウンレギュレーション
- 2014年6月号 Rho GTPase と活性酸素種: クロストークとフィードバック
- 2014年5月号 ミオシンのアセチル化はサルコメアの構造と機能を調節する
- 2014年4月号 リジンのアセチル化 - 多様な細胞プロセスの制御因子
- 2014年3月号 インテグリンを介したβ-アクチンの酸化還元制御: PDIの出現
- 2014年1/2月号 ダイニン: 一つのモーターが関わる複数の神経変性疾患
2013年
- 2013年11/12月号 ダイニン:チームとして強力に作用するモータータンパク質
- 2013年10月号 神経変性:Rhes、SUMO化、ハンチントン病
- 2013年9月号 モノユビキチン化:タンパク質調節のダイナミックなタグ
- 2013年8月号 Ras及びRhoのプレニル化による翻訳後修飾:癌創薬における役割
- 2013年7月号 アクチンが引き起こす膜突起による浸潤:コルタクチン
- 2013年6月号 アクチン修飾と細胞骨格
- 2013年5月号 微小管内部の実体
- 2013年4月号 神経変性におけるTauの多面性
- 2013年3月号 蛍光フィブロネクチンタンパク質を用いた特発性肺線維症の創薬
- 2013年1/2月号 樹状突起棘:発生におけるArf6の役割
2012年
- 2012年11/12月号 ミオシンの小分子モジュレーター
- 2012年10月号 Rhoファミリーパスウェイのユビキチン化と制御
- 2012年9月号 神経変性におけるRac1 GTPaseの機能
- 2012年8月号 上皮間葉転換(EMT)とRhoファミリー低分子量G-タンパク質の関与
- 2012年7月号 チューブリンの多重修飾:グルタミル化とグリシル化
- 2012年6月号 細胞接着のフィブロネクチン制御と原線維形成
- 2012年5月号 アクチン酸化サイクルの機能
- 2012年4月号 トラフィッキング:ArfとCdc42/Racの結合
- 2012年3月号 G-LISAを用いた心臓研究: 糖尿病性心筋症におけるRho経路に関する研究
- 2012年1月/2月号 FtsZ: 新たな抗生剤の標的となるチュ−ブリンホモログ
商品は「研究用試薬」です。人や動物の医療用・臨床診断用・食品用としては使用しないように、十分ご注意ください。
※ 表示価格について














