





















 このページを印刷する
このページを印刷する
記事ID : 17779
Tau の翻訳後修飾: アルツハイマー病の治療標的 CYTOSKELETON NEWS 2017年3月号
Tau の翻訳後修飾: アルツハイマー病の治療標的
はじめに
世界中で4,700万人を超える人々が認知症と診断されており、その大部分はアルツハイマー病(AD: Alzheimer’s disease)に起因します。神経変性疾患であるアルツハイマー病に関わるコストは、社会的負担を除くと、世界の国内総生産の 1.09% になります1。アルツハイマー病の病態生理学的な特徴として、運動や言語、認知の欠損をもたらす重度の認知障害が挙げられます。また、分子レベルでの神経病理学的な特徴には、Βアミロイド斑、および、2本の高リン酸化 Tau タンパク質のらせん状線維からなる神経原線維変化(NFT: neurofibrillary tangle)の形成が含まれます。本稿では、翻訳後修飾(PTM)による Tau の機構的制御と、Tau の翻訳後修飾の調節(図1)に基づく新規アルツハイマー病治療法の開発に注目します2。
Tau のリン酸化
脳脊髄液(CSF: cerebral spinal fluid)中の総 Tau 量の増加は、アルツハイマー病やその他の神経変性疾患と関連します。従って、総 Tau 量は、アルツハイマー病に特異的なマーカーとしてではなく、ニューロンの損傷および変性のマーカーとして定義されています3。反対に、脳脊髄液(CSF)中の T181 がリン酸化された tau 量の増加は、アルツハイマー病のみに関連することから、アルツハイマー病に特異的な数少ないバイオマーカーの1つになっています。神経原線維変化(NFT)は、死後にアルツハイマー病の病状を確認するために日常的に用いられており、近年の研究結果から、アルツハイマー病の Braak stage 初期に tau オリゴマーを検出できることが示唆されています。さらに、NFT の成熟と分布は、アルツハイマー病の認知機能低下に相関することから、アルツハイマー病の発病において神経原線維変化(NFT)が重要な役割を果たすことが示唆されます4。
翻訳後修飾の1つであるリン酸化により、Tau と微小管との生理的な相互作用が調節されます4。しかし、アルツハイマー病では、Tau が過剰にリン酸化されており、このリン酸化が Tau の誤った局在化、機能不全、凝集、神経原線維変化(NFT)形成を推進し、調節する機構となります5,6。Tau の過剰リン酸化は、45 個もの残基で生じており、その中には通常の Tau リン酸化部位とは異なる残基も含まれています3,6。Tau の過剰リン酸化は、cAMP 依存性プロテインキナーゼ、 c-Jun N末端キナーゼ3(JNK3)、グリコーゲン合成酵素キナーゼ3Β(GSK3B)、サイクリン依存性キナーゼ5(CDK5)などの数種類のキナーゼによって調節されます2。JNK3、CDK5、GSK3B 阻害剤は全て、in vitro および動物モデルにおいて神経保護特性を示します。GSK3B 阻害剤は、臨床試験が行われましたが、有意な効果が認められなかったため試験が終了しています2,7。しかし、脳脊髄液(CSF)中でCDK5 と共に増加するJNK3の阻害は、現在も治療法の候補となっており、詳細な検討が行われています。Tau の過剰リン酸化を調節する他の方法の一つに、Tau を脱リン酸化するホスファターゼの活性化があります。開発中のプロテインホスファターゼ2(PP2A)のアゴニストである亜セレン酸ナトリウムは、アルツハイマー病モデルマウスにおいて認知機能を改善することが示されています8,9。
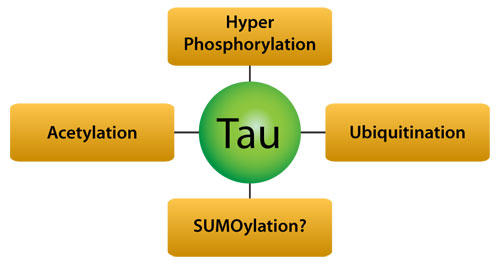
図1 Tau の翻訳後修飾はアルツハイマー病治療の標的となっている
Tau のユビキチン化とSUMO化
ユビキチン化とSUMO化(どちらも翻訳後修飾)もまた、Tau 活性および神経原線維変化(NFT)形成の重要な調節因子であることが同定されています。ユビキチンリガーゼである C-terminus of Hsc70-interacting protein(CHIP)は、タウオパチーモデルマウスにおいて、神経原線維変化(NFT)形成に対して有意な保護作用を示します。この知見は、アルツハイマー病脳において CHIP と病原性 Tau が逆の関係にあることを補完し、ユビキチン化が凝集 Tau の重要な除去機構である証拠となります10。近年行われた Luo らによる研究から、Tau の過剰リン酸化と Tau の SUMO化の間に重要なクロストークが確認され、どちらか一方が修飾されると他方の修飾が増加しました11。さらに、Tau の SUMO化は、Tau のポリユビキチン化と続いて起こる分解を防ぎ、凝集を引き起こす可能性があります。この研究から、Tau の様々な翻訳後修飾の間で重要なクロストークが起こっていることが強く示され、Tau の過剰リン酸化を最終的に調節するためには、翻訳後修飾の代替となる調節機構が標的になる可能性があります。実際に、過剰リン酸化とクロストークする翻訳後修飾(PTM)は、ユビキチン化やSUMO化に限定されていません。Tau の過剰リン酸化とグリコシル化との間のクロストークは、調節機構としてよく知られています12,13。
Tau のアセチル化
近年、アルツハイマー病の Braak stage 初期に増加する Tau の翻訳後修飾としてアセチル化が同定され、in vitro において過剰リン酸化された Tau の量と Tau の凝集を正に調節することが示されました14,15。脱アセチル化酵素 SIRT 1 の欠失により Tau のアセチル化が増加し、ポリユビキチン化と続いて起こるタンパク質のターンオーバーが抑制されることが示され、Tau のアセチル化がアルツハイマー病の進行を促進することを示すさらなる証拠となります14。これらの知見は、Tau の調節において翻訳後修飾のクロストークが重要であることを強く示しています。Min らは、これらの研究を基に、Tau のアセチル化に特異的で重要なリジン残基 K174 を同定し、Tau アセチル化の調節因子がリジンアセチルフェラーゼ P300 であることを明らかにしました16。K174 は、アルツハイマー病の Braak stage 初期および後期にアセチル化されます。重要なことに、P300 活性を低下させる医療用医薬品であるサルサラートは、タウオパチーモデルマウスにおいて、Tau を介した記憶障害と海馬の委縮を元に戻しました。さらに、サルサレートを、K174Q(リジン残基がアセチル化された Tau を模倣)を発現するニューロンに用いた場合は、総 Tau 量、リン酸化 Tau 量、海馬の委縮に変化が見られず、有意な効果は認められませんでした。このような変異体を用いた研究結果から、アルツハイマー病の進行においてアセチル化 Tau が重要な役割を果たすというさらなる証拠が得られています。この医薬品はすでにFDAの承認を受けていることから、アルツハイマー病患者の治療においても同じ効果が得られるのかどうかが注目されています。
まとめ
新たなアルツハイマー病治療の研究や創薬には、翻訳後修飾による Tau の調節を理解するだけではなく、癌、心血管、代謝、アルツハイマー病以外の神経疾患の多くの異常なタンパク質で、翻訳後修飾の調節機能が損なわれていることを考慮する必要があります17-19。例えば、チロシンキナーゼ受容体は癌において調節が解除される場合が多く、癌治療薬の中には、受容体の能力を調節することで下流の翻訳後修飾によるシグナル伝達制御を誘導するものがあります20。異常なタンパク質に対する新しい翻訳後修飾を同定することで、標的を絞った効果的な治療法の開発に利用できる可能性があります。さらに、翻訳後修飾のクロストークは、標的タンパク質の機能を調節するための基本的なメカニズムです。Cytoskeleton社では、標的タンパク質に対する新たな翻訳後修飾を同定するためのツールをご用意しており、標的タンパク質の調節機構の研究にご利用いただけます。Signal Seeker™ キットは、標的タンパク質の様々な翻訳後修飾(PTM)の内在性レベルを、高感度かつ定量的に測定することができます。
参考文献
- Prince M. et al. 2015. World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost & Trends; Alzheimer's Disease International: London, UK.
- Folch J. et al. 2016. Current Research Therapeutic Strategies for Alzheimer's Disease Treatment. Neural Plast.2016, 8501693.
- Russell C.L. et al. 2014. Post-translational modifications in Alzheimer's disease and the potential for new biomarkers. J. Alzheimers Dis. 41, 345-64.
- Simic G. et al. 2016. Tau protein hyperphosphorylation and aggregation in Alzheimer's disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 6, 6.
- Hoover B.R. et al. 2010. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 68, 1067-81.
- Hanger D.P. et al. 2009. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112-119.
- Lovestone S. et al. 2015. A phase II trial of tideglusib in Alzheimer's disease. J. Alzheimers Dis. 45, 75-88.
- Zhang Y. et al. 2014. Silencing [Formula: see text] Rescues Tau Pathologies and Memory Deficits through Rescuing PP2A and Inhibiting GSK-3beta Signaling in Human Tau Transgenic Mice. Front. Aging Neurosci. 6,123.
- van Eersel J. et al. 2010. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proc. Natl. Acad. Sci. USA. 107, 13888-93.
- Sahara N. et al. 2005. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 94,1254-1263.
- Luo H.B. et al. 2014. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA. 111, 16586-16591.
- Liu F. et al. 2004. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 101, 10804-10809.
- Lefebvre T. et al. 2003. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins--a role in nuclear localization. Biochim. Biophys. Acta. 1619, 167-76.
- Min S.W. et al. 2010. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 67, 953-966.
- Cohen T.J. et al. 2011. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2, 252.
- Min S.W. et al. 2015. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154-1162.
- Liddy K.A. et al. 2013. Functional decorations: post-translational modifications and heart disease delineated by targeted proteomics. Genome Med. 5, 20.
- Kim M.Y. et al. 2012. Role of transcription factor modifications in the pathogenesis of insulin resistance. Exp. Diabetes Res. 2012, 716425.
- Margolin D.H. et al. 2013. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 368, 1992-2003.
- Sullivan I. & Planchard D. 2016. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front. Med. (Lausanne). 3, 76.
Signal Seeker™ キット
[商品詳細]
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 | 在庫 |
|---|---|---|---|---|---|
Signal-SeekerTM Phosphotyrosine Detection Kit |
CYT | BK160 | 30 ASSAY |
CYT社 BK160L 1 を参照 |
なし |
| Signal-SeekerTM SUMOylation Detection Kit |
CYT | BK162 | 30 ASSAY |
CYT社 BK162L 1 を参照 |
なし |
| Signal-SeekerTM Ubiquitin Detection Kit |
CYT | BK161 | 30 ASSAY |
CYT社 BK161L 1 を参照 |
なし |
アクチン Biochem™ キット
[商品詳細]
- SUMO-2/3 アフィニティービーズ
- 免疫沈降(IP)によるSUMO化タンパク質の濃縮に最適
| 品名 | メーカー | 品番 | 包装 | 希望販売価格 | 在庫 |
|---|---|---|---|---|---|
| Anti Phosphotyrosine Affinity Beads, (Mouse) , 27B10.4 |
CYT | APY03-BEADS | 4*300 UL |
CYT社 APY03BEADS 4*330 を参照 |
なし |
| Anti SUMO-2/3 Affinity Beads, (Mouse) , 11G2 |
CYT | ASM24-BEADS | 800 UL [2 x 400 μl] |
CYT社 ASM24BEADSA 2*400 を参照 |
なし |
| Control for Ippt IgG beads |
CYT | CIG01-BEADS | 10 ASSAY |
CYT社 CIG01BEADS10 10 を参照 |
なし |
■ CYTOSKELETON NEWS バックナンバー
- 2020年10月号 紡錘体 - 可視化に向けた新規ツール
- 2020年8月号 細胞膜染色用蛍光プローブ
- 2020年4月号 生細胞におけるF-アクチンプローブ
- 2020年3月号 コロナウイルスと細胞骨格
- 2020年2月号 タウ(Tau)の将来性をMapping
- 2020年1月号 Rho GTPaseによる細胞遊走制御
- 2019年12月号 表現型プロファイリング:アクチンに焦点を当てたがん治療
- 2019年11月号 チューブリンの過剰グルタミル化、ミトコンドリア、神経変性
- 2019年9月号 細胞運動性を制御するために相互作用する膜張力とアクチン細胞骨格
- 2019年8月号 Rac1B、がん、およびRac1
- 2019年7月号 Rhoファミリー GTPases、神経可塑性、およびうつ状態
- 2019年6月号 アクチンメチオニン酸化: 動的制御の次の段階
- 2019年5月号 ミクログリアと神経変性疾患
- 2019年2月号 生細胞画像化に対するCNS疾患や障害
2018年
- 2018年12月号 アクチン細胞骨格とメカノトランスダクション(機械的シグナル伝達)
- 2018年11月号 軸索再生と細胞骨格
- 2018年10月号 ニューロンにおける微小管と極性
- 2018年8月号 Rab GTPase と 神経変性
- 2018年7月号 SUMO レスリング: バランスが全て
- 2018年6月号 なぜ K-Ras は発がん特異性を示すのか?
- 2018年5月号 治療標的としてのユビキチンプロテアソームシステム:チューブリンは関与するか?
- 2018年4月号 RhoファミリーGEFと樹状突起スパインの構造的可塑性
- 2018年3月号 βカテニンとTFC/LEF-1の翻訳後修飾による標準的なWntシグナル制御
- 2018年2月号 がん抑制遺伝子p53の翻訳後修飾による機能の調整
- 2018年1月号 自閉スペクトラム症におけるGEF Trioの役割
2017年
- 2017年12月号 プロフィリン: アクチン結合タンパク質の多機能な役割
- 2017年11月号 ミトコンドリアにおけるアセチル化:新たな考え方と治療への応用の可能性
- 2017年9月号 翻訳後修飾のアセチル化による微小管の安定化
- 2017年8月号 神経軸索におけるアクチンリングを基盤とした周期的膜骨格(PMS)
- 2017年7月号 E3ユビキチンリガーゼMdm2によるがん抑制遺伝子p53の翻訳後制御
- 2017年6月号 多能性幹細胞(PSC)での転写因子による翻訳後制御
- 2017年5月号 Arf6 GEF と癌細胞の浸潤・転移
- 2017年4月号 PTEN(Phosphatase and Tensin Homolog)による翻訳後制御
- 2017年3月号 Tau の翻訳後修飾: アルツハイマー病の治療標的
- 2017年2月号 樹状細胞の移動におけるアクチン結合タンパク質とF-アクチン
2016年
- 2016年11月/12月号 GEF を介した GTPase シグナル伝達の低分子阻害剤
- 2016年9月号 FtsZ タンパク質: 抗菌薬の新規ターゲット
- 2016年7月号 翻訳後修飾(PTM)は心臓病において細胞骨格タンパク質を調節する
- 2016年6月号 モータータンパク質キネシンと神経変性
- 2016年5月号 チロシンリン酸化は Rhoファミリー GTPase 活性を調節する
- 2016年4月号 Rac1と糖尿病: ポジティブな役割とネガティブな役割
- 2016年3月号 SUMO化: 細胞骨格タンパク質の機能を調節するレギュレーター
- 2016年1月/2月号 ビメンチン中間径フィラメント: リン酸化による調節
2015年
- 2015年8月号 タンパク質調節に不可欠な翻訳後修飾
- 2015年7月号 アクチン細胞骨格のライブセルイメージング
- 2015年6月号 有糸分裂に関わるタンパク質のSUMO化: 局在と機能
- 2015年5月号 Ras 癌の治療: 5つの有望なターゲット
- 2015年4月号 Ras 依存性の癌で注目される YAP1
- 2015年3月号 増刊号 統合失調症において遺伝子変異により誘導されるアクチン依存のシナプスの変化
- 2015年3月号 Ral GTPase を調節する翻訳後修飾
- 2015年1月/2月号 RhoA のリン酸化はシグナル伝達を調節する
- 2015年1月号 増刊号 微小管を不安定化する suprafenacine: 新規抗癌剤のリード化合物としての可能性
2014年
- 2014年12月号 増刊号 RhoA は心筋細胞におけるアクチン細胞骨格の再構成とグルコース取り込みを仲介する
- 2014年11月号 増刊号 樹状突起の形態形成: ドーパミンD1受容体 および Rho ファミリー GTPase による制御
- 2014年11月/12月号 GTPase 活性化アッセイ: アイソフォームの検出
- 2014年10月号 アルギニンの正電荷を消失させるシトルリン化
- 2014年9月号 キネシンサブドメインの探索
- 2014年9月号 増刊号 アクチン結合タンパク質コフィリンの S-ニトロシル化: 細胞移動に対する影響
- 2014年8月号 増刊号 原発性硬化性胆管炎における N-Ras 発現および活性
- 2014年8月号 SUMO化: 細胞骨格タンパク質を標的とした翻訳後修飾
- 2014年7月号 Sos/K-Ras 結合を介して Ras シグナル伝達を制御する新しい低分子阻害剤
- 2014年6月号 増刊号 頭頸部扁平上皮癌における microRNA-138 による RhoC のダウンレギュレーション
- 2014年6月号 Rho GTPase と活性酸素種: クロストークとフィードバック
- 2014年5月号 ミオシンのアセチル化はサルコメアの構造と機能を調節する
- 2014年4月号 リジンのアセチル化 - 多様な細胞プロセスの制御因子
- 2014年3月号 インテグリンを介したβ-アクチンの酸化還元制御: PDIの出現
- 2014年1/2月号 ダイニン: 一つのモーターが関わる複数の神経変性疾患
2013年
- 2013年11/12月号 ダイニン:チームとして強力に作用するモータータンパク質
- 2013年10月号 神経変性:Rhes、SUMO化、ハンチントン病
- 2013年9月号 モノユビキチン化:タンパク質調節のダイナミックなタグ
- 2013年8月号 Ras及びRhoのプレニル化による翻訳後修飾:癌創薬における役割
- 2013年7月号 アクチンが引き起こす膜突起による浸潤:コルタクチン
- 2013年6月号 アクチン修飾と細胞骨格
- 2013年5月号 微小管内部の実体
- 2013年4月号 神経変性におけるTauの多面性
- 2013年3月号 蛍光フィブロネクチンタンパク質を用いた特発性肺線維症の創薬
- 2013年1/2月号 樹状突起棘:発生におけるArf6の役割
2012年
- 2012年11/12月号 ミオシンの小分子モジュレーター
- 2012年10月号 Rhoファミリーパスウェイのユビキチン化と制御
- 2012年9月号 神経変性におけるRac1 GTPaseの機能
- 2012年8月号 上皮間葉転換(EMT)とRhoファミリー低分子量G-タンパク質の関与
- 2012年7月号 チューブリンの多重修飾:グルタミル化とグリシル化
- 2012年6月号 細胞接着のフィブロネクチン制御と原線維形成
- 2012年5月号 アクチン酸化サイクルの機能
- 2012年4月号 トラフィッキング:ArfとCdc42/Racの結合
- 2012年3月号 G-LISAを用いた心臓研究: 糖尿病性心筋症におけるRho経路に関する研究
- 2012年1月/2月号 FtsZ: 新たな抗生剤の標的となるチュ−ブリンホモログ
商品は「研究用試薬」です。人や動物の医療用・臨床診断用・食品用としては使用しないように、十分ご注意ください。
※ 表示価格について














